hello,大家好,随着单细胞数据越来越火,我们需要的基础知识也越来越多,也越来越精细,今天给大家分享一些分析的细节知识,所谓细节决定成败,也许自己的数据分析的不好,就是细节没有做好,话不多说,进入正题
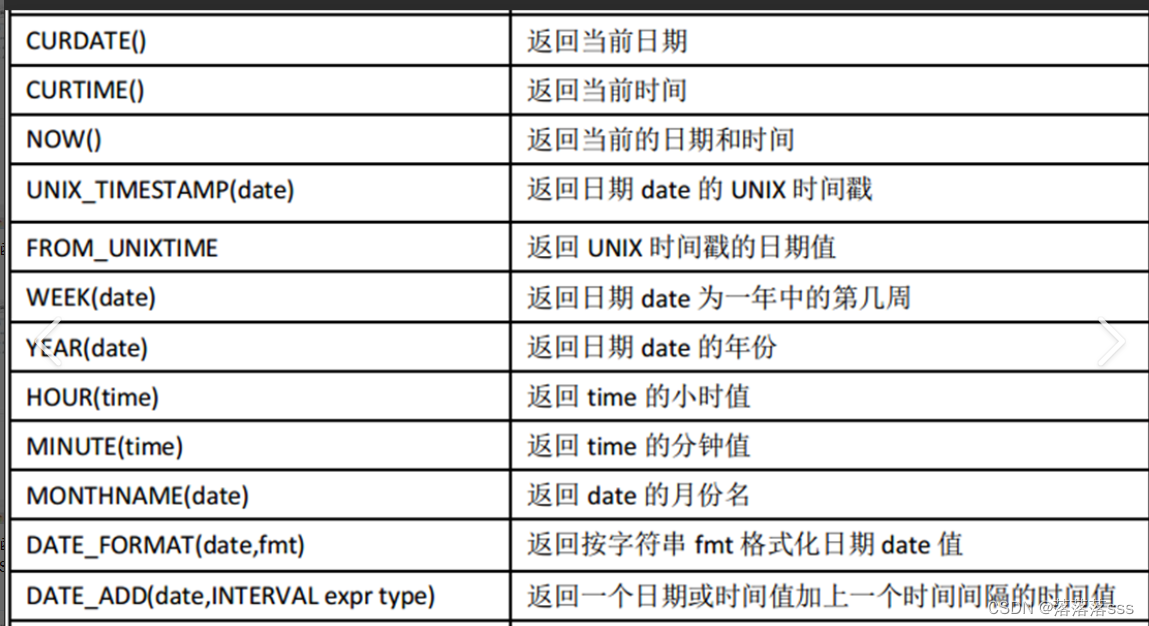
首先第一个问题,空油滴的问题,也就是没有捕获到细胞的GEMs,如何识别呢?我们先来看下面的图:

这个图大家应该都很熟悉吧,(不知道的自动面壁去😄),目前的cellranger版本对于空油滴(背景)的识别很粗糙,UMI数量在500以上的全部保留,但是对保留的细胞中,有一部分是否为细胞提供了概率值,当然我们一般都当作细胞来处理,进行下游分析了,很明显,这个方法很粗糙,这个地方需要留心,当然,下游分析无论是Seurat还是scanpy都不会对背景再次的识别,只是去除低质量的细胞,那么很多时候,我们需要一个科学的方法来帮助我们去掉“背景的细胞”。
Raw UMI count matrices were imported into R for further processing. For each scRNA-Seq sample, cell calling was performed using ‘emptyDrops’ function from DropletUtils (version 1.4.3) on the full raw count matrices in order to distinguish cells from empty droplets containing only ambient RNA(采用一定的算法,也就是软件辅助去除空油滴,而不再是简单粗暴的去除). Raw count matrices were corrected for Illumina index swapping using ‘swapped-Drops’ . This identified 140,264 non-empty droplets across all single cell pools.这个分析来自于文章Spatiotemporal analysis of human intestinal development at single-cell resolution,2021年初发表于Cell,值得借鉴,其中用到的软件DropletUtils,文章在EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data,2019年发表于Genome Biology,影响因子11分,软件在DropletUtils,代码相对简单,但是背后的算法原理,大家需要认真学习一下了。
第二个问题,低质量细胞的过滤,其实关于10X单细胞数据过滤的问题,在我分享的文章10X单细胞(10X空间转录组)数据分析之细胞过滤那些事,大家感兴趣可以看一下,而今天我们需要更进一步了。
droplet barcodes for which a high percentage of total UMIs originated from mitochondrial RNAs were filtered out, as well as low total UMI count barcodes. These thresholds were derived individually for cells within each compartment following an initial clustering solution of all cells by examining and thresholding empirical distributions within each compartment, as total RNA content (notably higher in endothelial and myeloid cell populations) and mitochondrial RNA content (notably higher in epithelial cells) are highly cell type dependent.这个地方一定要注意,我已经标红了,线粒体和低UMI的细胞过滤不是一刀切的过程,每种细胞类型的过滤阈值依据最初的聚类方案做出的,换句话讲,每个细胞类型的线粒体过滤阈值都不一样,所以在过滤的时候,每种细胞类型都有一个阈值,以此来过滤细胞,怎么样,是不是不符合你最初的认识??如果有能力,这样做是最好。
第三个问题,非线性降维的问题
For each individual pool, Seurat (Butler et al., 2018) R package (version 3.1.5.9900) was used to normalize expression values for total UMI counts per cell. Highly variable genes were identified by fitting the mean-variance relationship and dimensionality reduction was performed using principal-component analysis. Scree plots were used to determine principal components to use for clustering analyses for each pool. Cells were then clustered using Louvain algorithm for modularity optimization using kNN graph as input. Cell clusters were visualized using UMAP algorithm (McInnes et al., 2018) with principal components as input and n.neighbors = 30, spread = 1 and min.dist = 0.1.
看起来很常规,但是我们需要注意这里 n.neighbors = 30, spread = 1 and min.dist = 0.1,我相信很多人在进行UMAP降维的时候都采用默认是,我们先来看看默认值是多少,n.neighbors = 30, spread = 1 and min.dist = 0.3,这三个参数的意义我们来看一下:
- n.neighbors: This determines the number of neighboring points used in local approximations of manifold structure. Larger values will result in more global structure being preserved at the loss of detailed local structure. In general this parameter should often be in the range 5 to 50.(跟流形学习有关,值越大更加保留全局结构而损失local结构,这里不详细展开聊了,在我分享的文章10X单细胞(10X空间转录组)降维分析之tSNE(算法基础知识)已经有过详细的介绍了)。
- spread:The effective scale of embedded points. In combination with min.dist this determines how clustered/clumped the embedded points are.(决定降维结构点)。
- min.dist:This controls how tightly the embedding is allowed compress points together. Larger values ensure embedded points are moreevenly distributed, while smaller values allow the algorithm to optimise more accurately with regard to local structure. Sensible values are in the range 0.001 to 0.5.(决定点降维的紧密程度,较大的值可确保嵌入点分布更均匀,而较小的值可使算法在局部结构方面进行更准确的优化。)。
这三个参数很重要,不过大家通常分析样本的时候默认参数就可以,但是在某个群细分的时候,参数就要根据自己的需要变化了。注意参数的意义。
第四个问题,批次矫正的问题
Cells from separate pools were merged and pool batch effect signal was corrected using harmony (version 1.0) algorithm(harmony矫正)。Merged cell clustering and visualization of cells from all pools was performed as before using Louvain and UMAP algorithms, using harmony dimensionality reduction as input instead of principal components. Merged pool clusters were compared with cell types obtained from individual pools to ensure cell type heterogeneity was not lost due to batch correction.
这个地方,个人认为最为关键,首先矫正采用的是harmony,这个可能大家都有过了解,甚至在用,但是看一看这里的做法,10X单细胞数据也要进行数据分析和定义,整合之后的分析结果要确保单样本分析得到的细胞类型没有因此矫正而丧失其异质性,这个地方很关键,也是我一直坚持的观点,大家一定要深入挖掘自己的数据,不可马虎大意。。
第五个问题,再分群问题
Merged cell data was then divided into compartments based on clustering analysis and marker gene expression, as outlined above. Cells from epithelial, endothelial, pericyte, muscularis, neural, fibroblast, immune, myofibroblast and mesothelial compartments were subset for further analysis. For each compartment, we carried out compartment-specific QC, batch correction and clustering analyses as described above.
注意了,多样本整合后的结果每种细胞类型再分群分析的时候,For each compartment, we carried out compartment-specific QC, batch correction and clustering analyses as described above。一定要注意,千万别用老式的那一套。
看到了吧,细节决定成败,这就是别人发Nature、Cell、Science,你只能发几分的原因,加油吧,路漫漫其修远兮,吾将上下而求索。
天行健,君子以自强不息