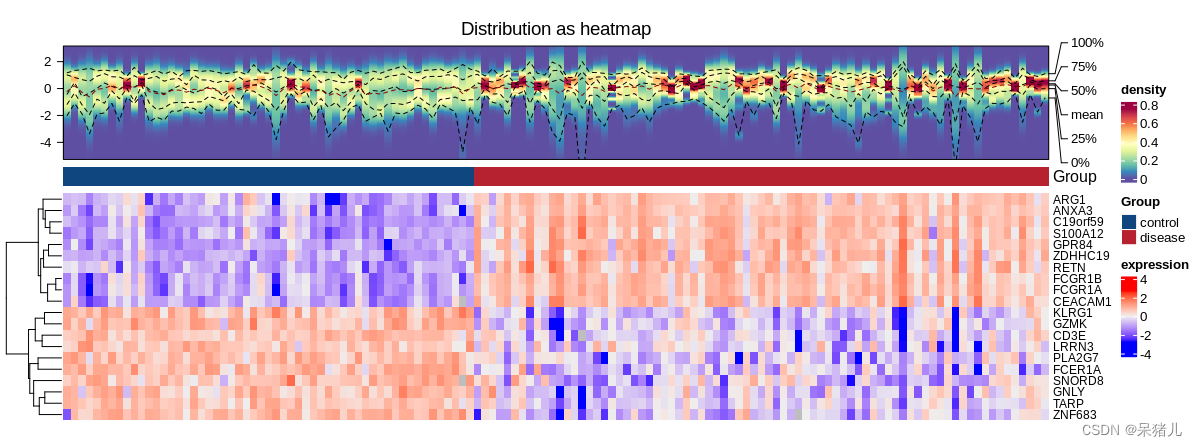
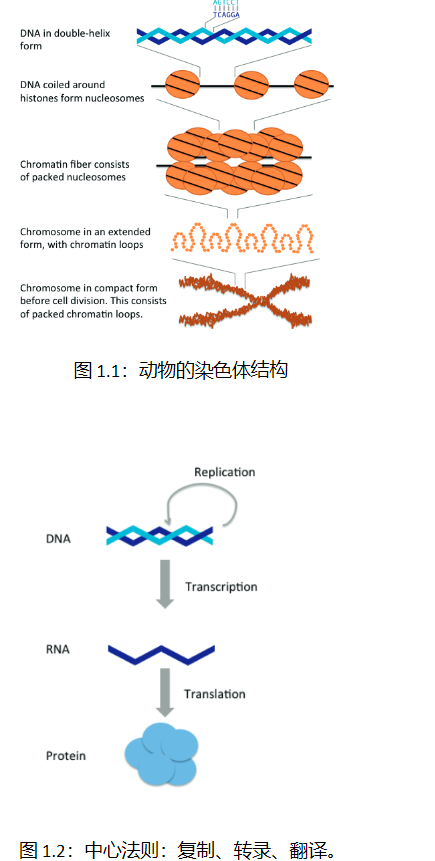
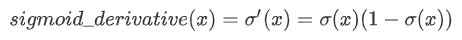生信中的差异分析是研究不同样本或条件下基因表达、蛋白质表达等差异的方法之一。下面介绍几种常用的差异分析方法:
基于微阵列的差异分析:使用基因芯片技术,将基因表达水平转化为信号强度,通过对比不同样本的信号强度来鉴定差异基因。常用的方法有t-检验、方差分析、秩和检验等。
RNA-seq差异分析:通过高通量测序技术获取基因组的转录本信息,可以定量测量基因表达水平。差异分析常采用基于计数的方法,如DESeq2、edgeR等,通过负二项分布模型或负二项模型来比较两组或多组样本的基因表达的差异。
蛋白质质谱差异分析:通过质谱技术测量蛋白质样本中的肽段,并通过鉴定和定量这些肽段来推断蛋白质的差异表达。常用的方法有t-检验、ANOVA、Kruskal-Wallis检验等。
DNA甲基化差异分析:通过测量DNA中的甲基化水平差异来研究不同样本间的DNA甲基化变化。常用的方法有DMRcaller、methylKit等,可以鉴定差异甲基化位点及差异甲基化区域。
miRNA差异分析:通过测量miRNA的表达水平来识别不同样本或条件下的miRNA差异。常用的方法有DESeq2、edgeR等。
这些差异分析方法在生信研究中发挥着重要作用,根据研究目的和数据类型的不同,选择合适的差异分析方法可以有效地鉴定差异基因或差异分子,并对生物学过程进行进一步的解析。
DESeq2和edgeR是两种常用的差异表达分析工具,用于分析RNA-Seq数据中基因的差异表达情况。它们基于统计模型来计算基因表达的差异,并提供了一系列的统计检验和可视化工具,帮助研究人员发现与不同生物条件相关的差异表达基因。
DESeq2是一种基于负二项分布的差异表达分析方法。该方法通过计算基因在样本间的差异表达,并考虑到测序数据的离散性和分布的特征。DESeq2使用了一个广义线性模型来描述基因表达量与不同条件之间的关系,并使用负二项分布来模拟测序数据的离散性。DESeq2通过对每个基因进行假设检验来确定差异表达的基因,并根据调整后的p值和折叠变化(fold change)来筛选显著差异的基因。
edgeR是另一种常用的差异表达分析方法,也适用于RNA-Seq数据的分析。与DESeq2类似,edgeR也考虑了RNA-Seq数据的离散性和分布的特征。edgeR使用了负二项分布来将测序数据建模,并采用一个广义线性模型来描述基因表达量与条件之间的关系。与DESeq2不同的是,edgeR将基因表达量归一化到每百万reads的标准化计数,然后进行基因表达的差异分析。edgeR通过计算每个基因的假设检验p值和折叠变化来确定差异表达的基因。
DESeq2和edgeR在差异表达分析中都得到了广泛的应用。它们都考虑了RNA-Seq数据的离散性和分布的特征,并提供了统计学上严格的假设检验方法。此外,这两种方法都提供了丰富的统计检验和可视化工具,用于帮助研究人员更好地理解差异表达的结果。选择DESeq2还是edgeR通常取决于研究人员对数据和方法的偏好,以及具体研究问题的要求。
DESeq2是一种用于RNA-Seq数据分析的R包,而edgeR则是另一种常用的RNA-Seq数据分析工具。
下面是一些使用DESeq2和edgeR的常见代码示例:
使用DESeq2进行差异表达分析:
- 安装和加载DESeq2包:
install.packages("DESeq2")
library("DESeq2")
- 读取表达矩阵数据和样本信息:
countMatrix <- read.csv("expression_data.csv", header = TRUE, row.names = 1)
sampleInfo <- read.csv("sample_info.csv", header = TRUE)
- 创建DESeq2对象和样本条件:
dds <- DESeqDataSetFromMatrix(countData = countMatrix, colData = sampleInfo, design = ~ condition)
- 样本间归一化和基因表达值估计:
dds <- DESeq(dds)
- 寻找差异表达基因:
res <- results(dds)
使用edgeR进行差异表达分析:
- 安装和加载edgeR包:
install.packages("edgeR")
library("edgeR")
- 读取表达矩阵数据和样本信息:
countMatrix <- read.csv("expression_data.csv", header = TRUE, row.names = 1)
sampleInfo <- read.csv("sample_info.csv", header = TRUE)
- 创建edgeR对象和样本条件:
dge <- DGEList(counts = countMatrix, group = sampleInfo$condition)
- 样本间归一化和基因表达值估计:
dge <- calcNormFactors(dge)
dge <- estimateCommonDisp(dge)
dge <- estimateTagwiseDisp(dge)
- 寻找差异表达基因:
fit <- glmFit(dge, design = model.matrix(~condition))
lrt <- glmLRT(fit)
topGenes <- topTags(lrt, n = 100)
以上代码仅为示例,实际分析中可能需要进行更多的数据预处理和结果解释。请根据具体需求和数据类型进行相应的参数设定和结果处理。

























![SCI 1区论文:Segment anything in medical images(MedSAM)[文献阅读]](https://img-blog.csdnimg.cn/img_convert/6d849a59d4d8126efd8cf9a830a7fe46.png)