> library("clusterProfiler")
> library("org.Hs.eg.db")
> library("enrichplot")
> library("ggplot2")
> library("org.Hs.eg.db")
#install.packages("GOplot")
> library(GOplot)
> rt=read.table("input.txt",sep="\t",check.names=F,header=T)
> genes=as.vector(rt[,1])
> entrezIDs <- mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA)
> entrezIDs <- as.character(entrezIDs)
> out=cbind(rt,entrezID=entrezIDs)
> write.table(out,file="GO-id.txt",sep="\t",quote=F,row.names=F)
> rt=read.table("GO-id.txt",sep="\t",header=T,check.names=F)
> rt=rt[is.na(rt[,"entrezID"])==F,]
> gene=rt$entrezID
> kk <- enrichGO(gene = gene,OrgDb = org.Hs.eg.db, pvalueCutoff =0.05, qvalueCutoff = 0.05,ont="all",readable =T)
> write.table(kk,file="GO.txt",sep="\t",quote=F,row.names = F)
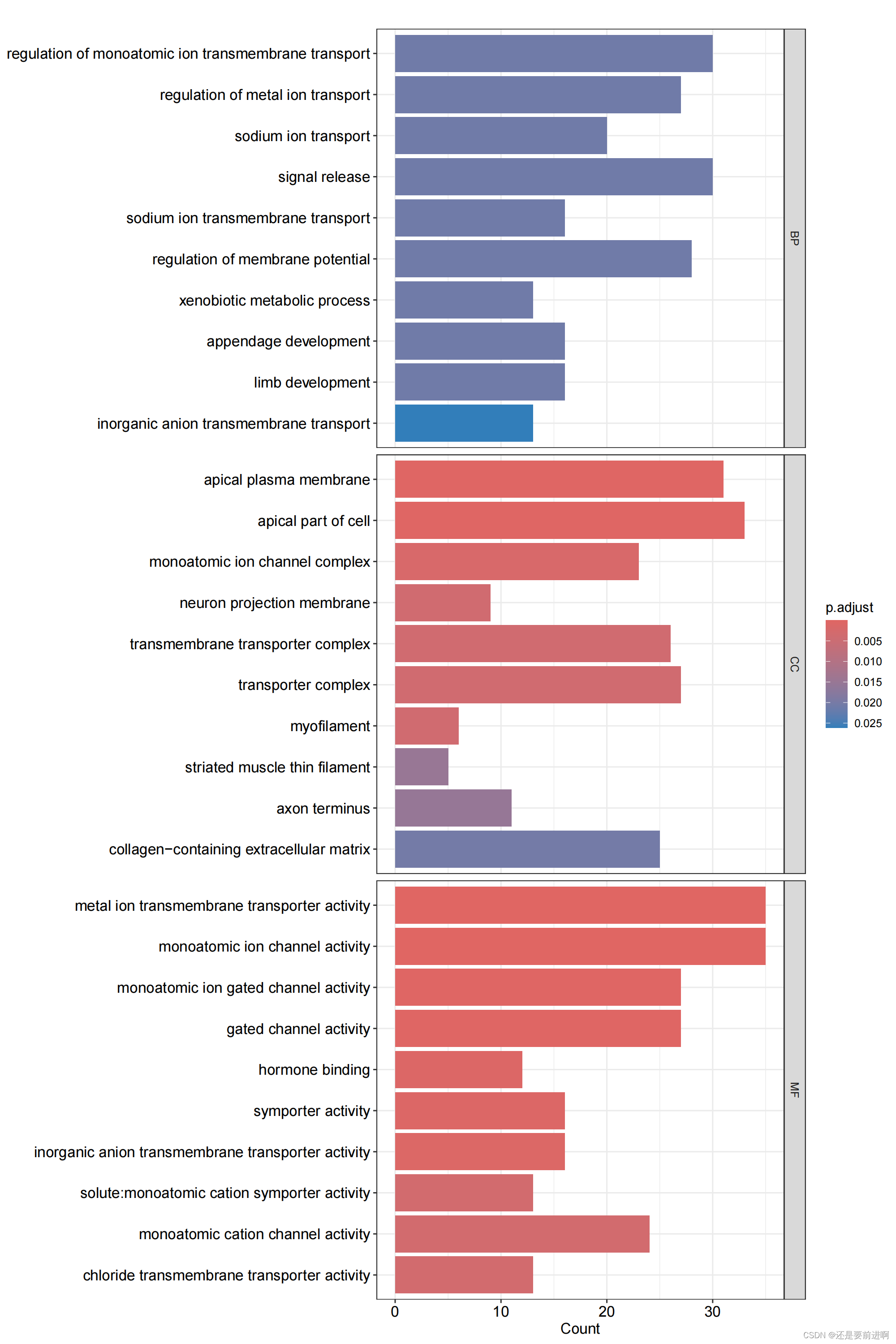
> pdf(file="GO-barplot.pdf",width = 10,height = 15)
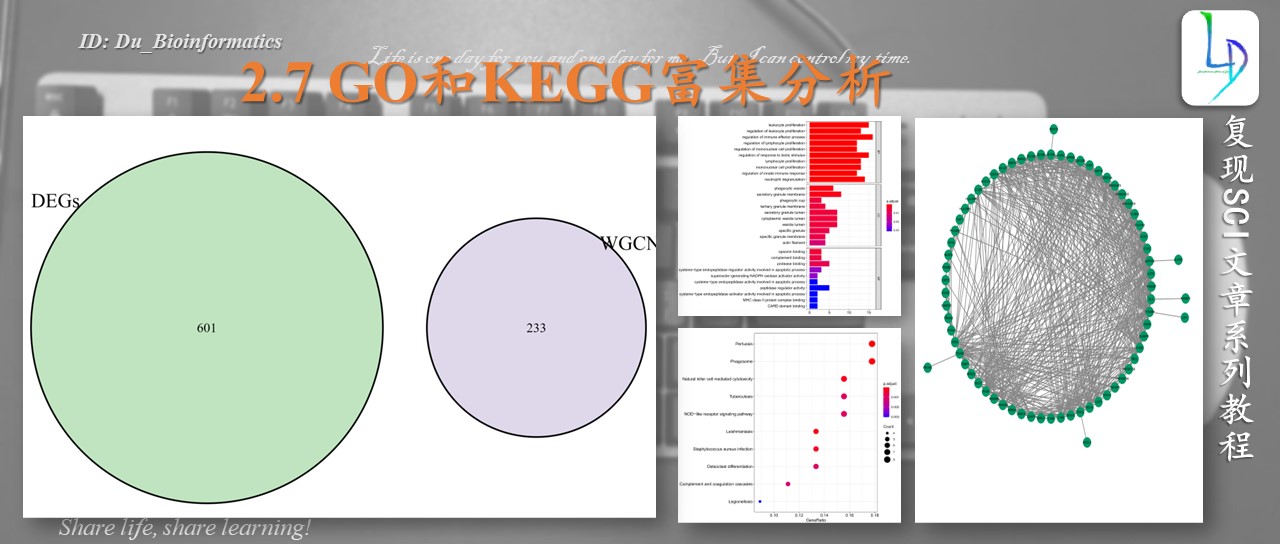
> barplot(kk, drop = TRUE, showCategory =10,label_format=100,split="ONTOLOGY") + facet_grid(ONTOLOGY~., scale='free')
> dev.off()
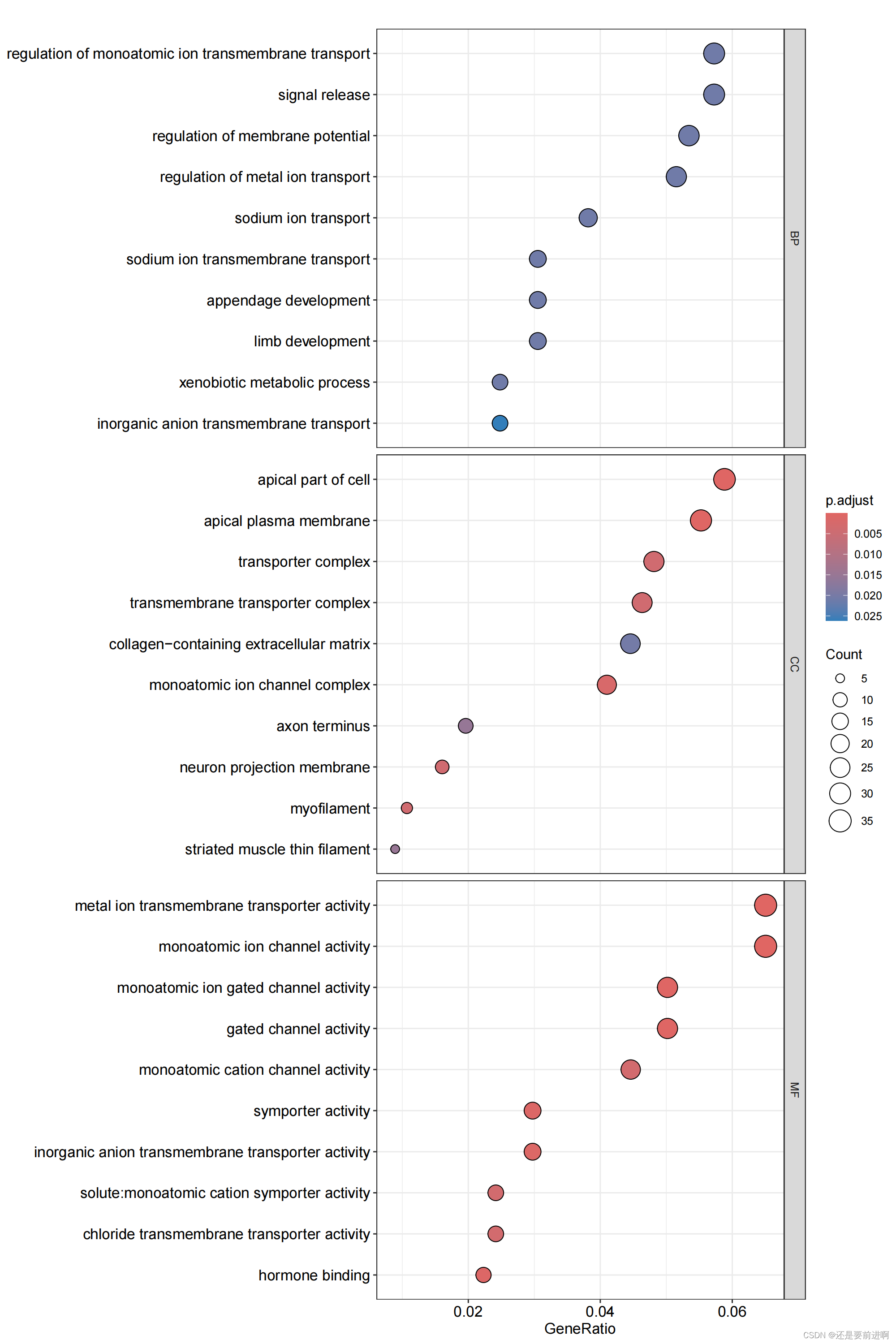
> pdf(file="GO-bubble.pdf",width = 10,height = 15)
> dotplot(kk,showCategory = 10,label_format=100,split="ONTOLOGY") + facet_grid(ONTOLOGY~., scale='free')
> dev.off()
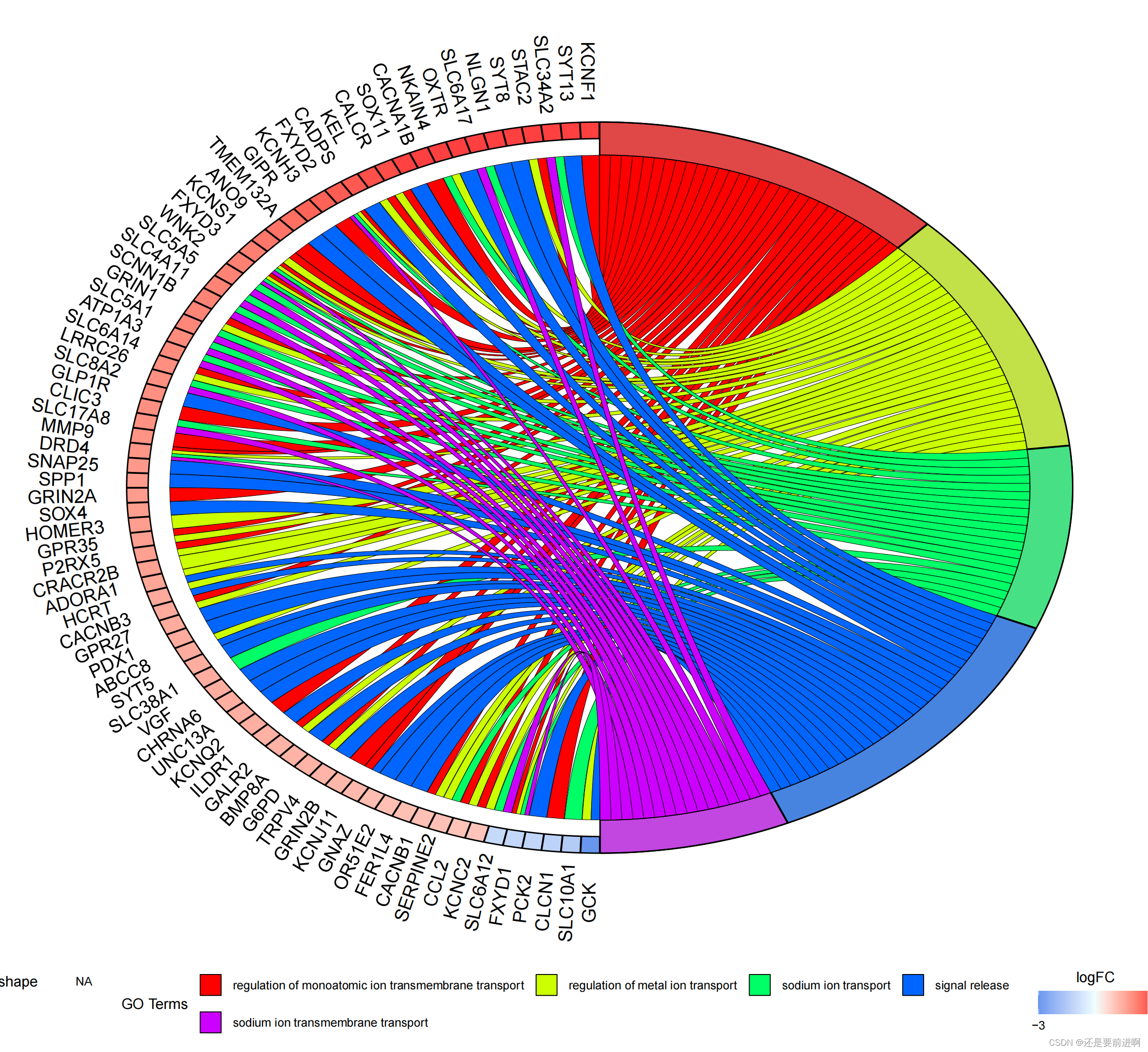
ego<-read.table("GO.txt",sep="\t",check.names=F,header=T)
go=data.frame(Category ="ALL",ID = ego$ID,Term = ego$Description, Genes = gsub("/", ", ", ego$geneID), adj_pval = ego$pvalue)
id.fc=rt
genelist <- data.frame(ID = id.fc$gene, logFC = id.fc$logFC)
row.names(genelist)=genelist[,1]
circ <- circle_dat(go, genelist)
termNum = 5
geneNum = nrow(genelist)
chord <- chord_dat(circ, genelist[1:geneNum,], go$Term[1:termNum])
pdf(file="GO_circ.pdf",width = 12,height = 11)
GOChord(chord,
space = 0.001,
gene.order = 'logFC',
gene.space = 0.25,
gene.size = 5,
border.size = 0.1,
process.label = 9)
dev.off()
> rt=read.table("input.txt",sep="\t",check.names=F,header=T)
> genes=as.vector(rt[,1])
> entrezIDs <- mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA)
> entrezIDs <- as.character(entrezIDs)
> out=cbind(rt,entrezID=entrezIDs)
> write.table(out,file="KEGG-id.txt",sep="\t",quote=F,row.names=F)
> rt=read.table("KEGG-id.txt",sep="\t",header=T,check.names=F)
> rt=rt[is.na(rt[,"entrezID"])==F,]
> gene=rt$entrezID
> kk <- enrichKEGG(gene = gene,keyType = "kegg",organism = "hsa", pvalueCutoff =0.05, qvalueCutoff =0.05, pAdjustMethod = "fdr")
> write.table(kk,file="KEGG.txt",sep="\t",quote=F,row.names = F)
> pdf(file="KEGG-barplot.pdf",width = 10,height = 13)
> barplot(kk, drop = TRUE, showCategory = 15,label_format=100)
> dev.off()
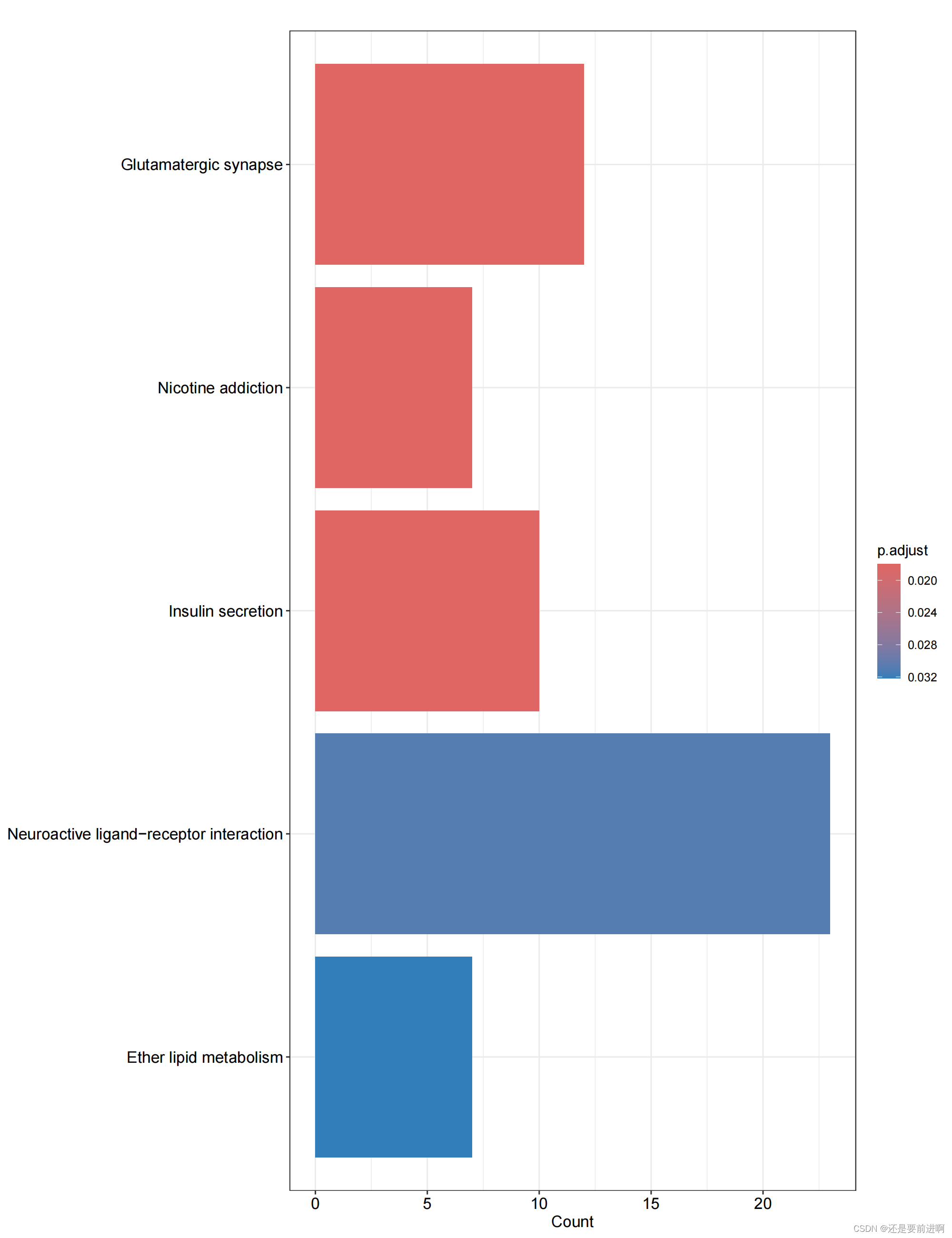
> pdf(file="KEGG-bubble.pdf",width = 10,height = 13)
> dotplot(kk, showCategory = 15,label_format=100)
> dev.off()
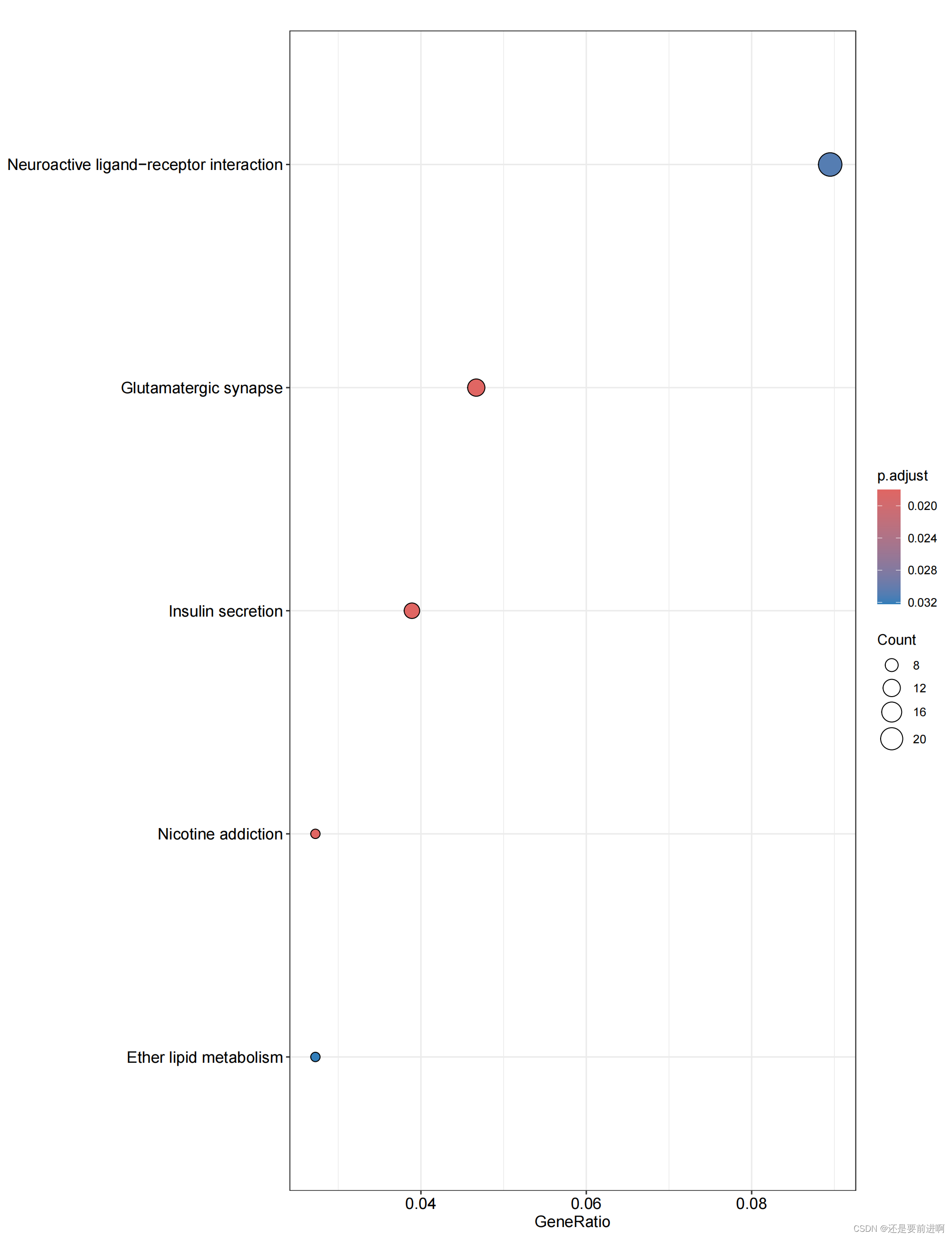
> ego<-read.table("KEGG.txt",sep="\t",check.names=F,header=T)
> go=data.frame(Category ="ALL",ID = ego$ID,Term = ego$Description, Genes = gsub("/", ", ", ego$geneID), adj_pval = ego$p.adjust)
> id.fc=rt
> genelist <- data.frame(ID = id.fc$entrezID, logFC = id.fc$logFC)
> row.names(genelist)=genelist[,1]
> row.names(rt)=rt[,3]
> circ <- circle_dat(go, genelist)
> termNum = 5
> geneNum = nrow(genelist)
> chord <- chord_dat(circ, genelist[1:geneNum,], go$Term[1:termNum])
> sameSample=intersect(row.names(chord), row.names(rt))
> rt=rt[sameSample,,drop=F]
> geneIDs=rt$gene
> row.names(chord)=geneIDs
> pdf(file="KEGG_circ.pdf",width = 12,height = 11)
> GOChord(chord,
space = 0.001,
gene.order = 'logFC',
gene.space = 0.25,
gene.size = 5,
border.size = 0.1,
process.label = 9)
> dev.off()
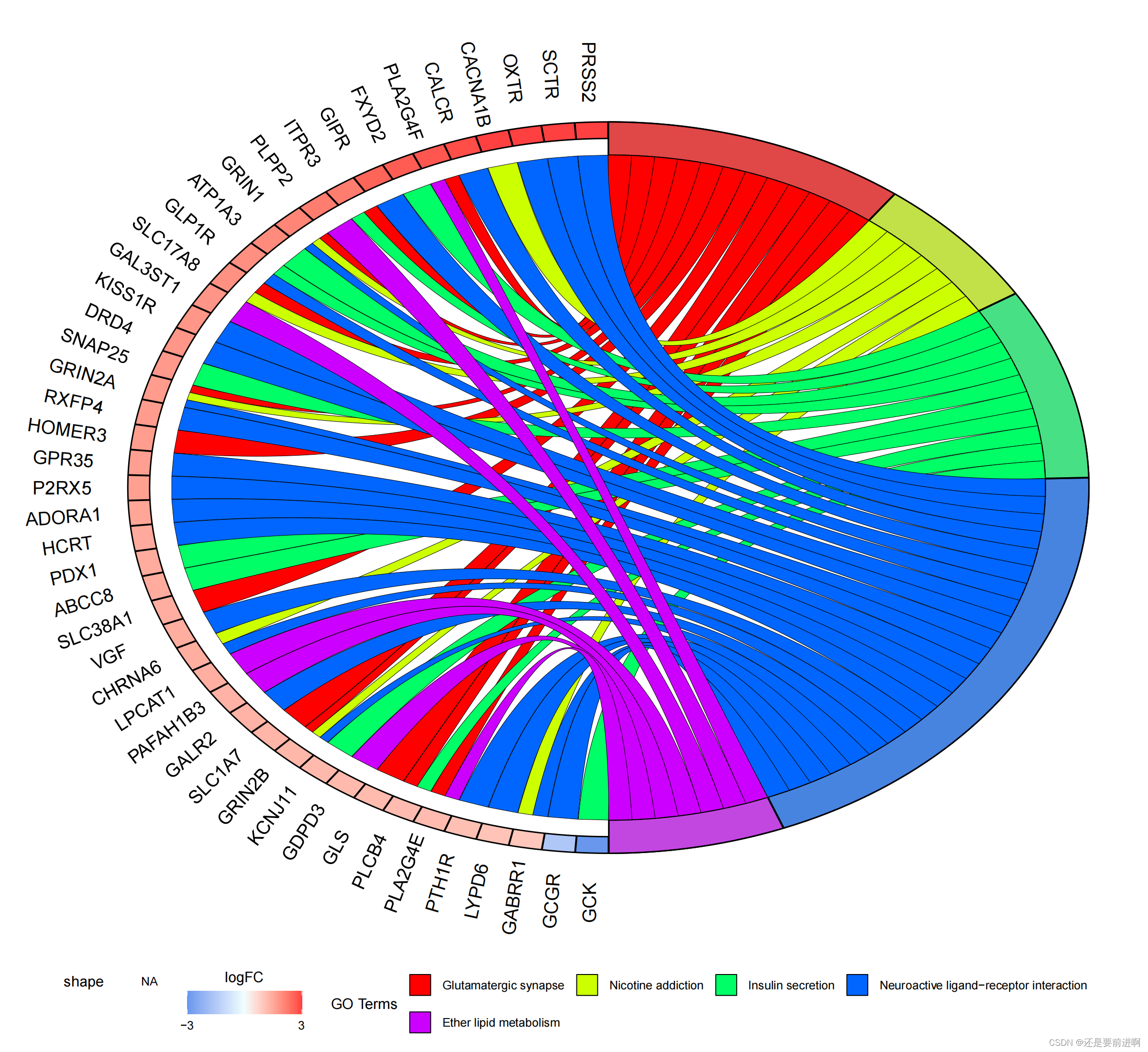
学习交流一下