假如有bulk-RNA测序的数据:TH1,TH2,TH3三个重复(实验组),TW1,TW2,TW3三个重复(对照组)
- 准备工作
需要安装的软件(如FastQC、Trimmomatic、HISAT2、StringTie、samtools)
conda install -c bioconda fastqc
conda install trimmomatic
conda install -c bioconda stringtie
conda install -c bioconda hisat2
conda install samtools
conda install -c bioconda picard
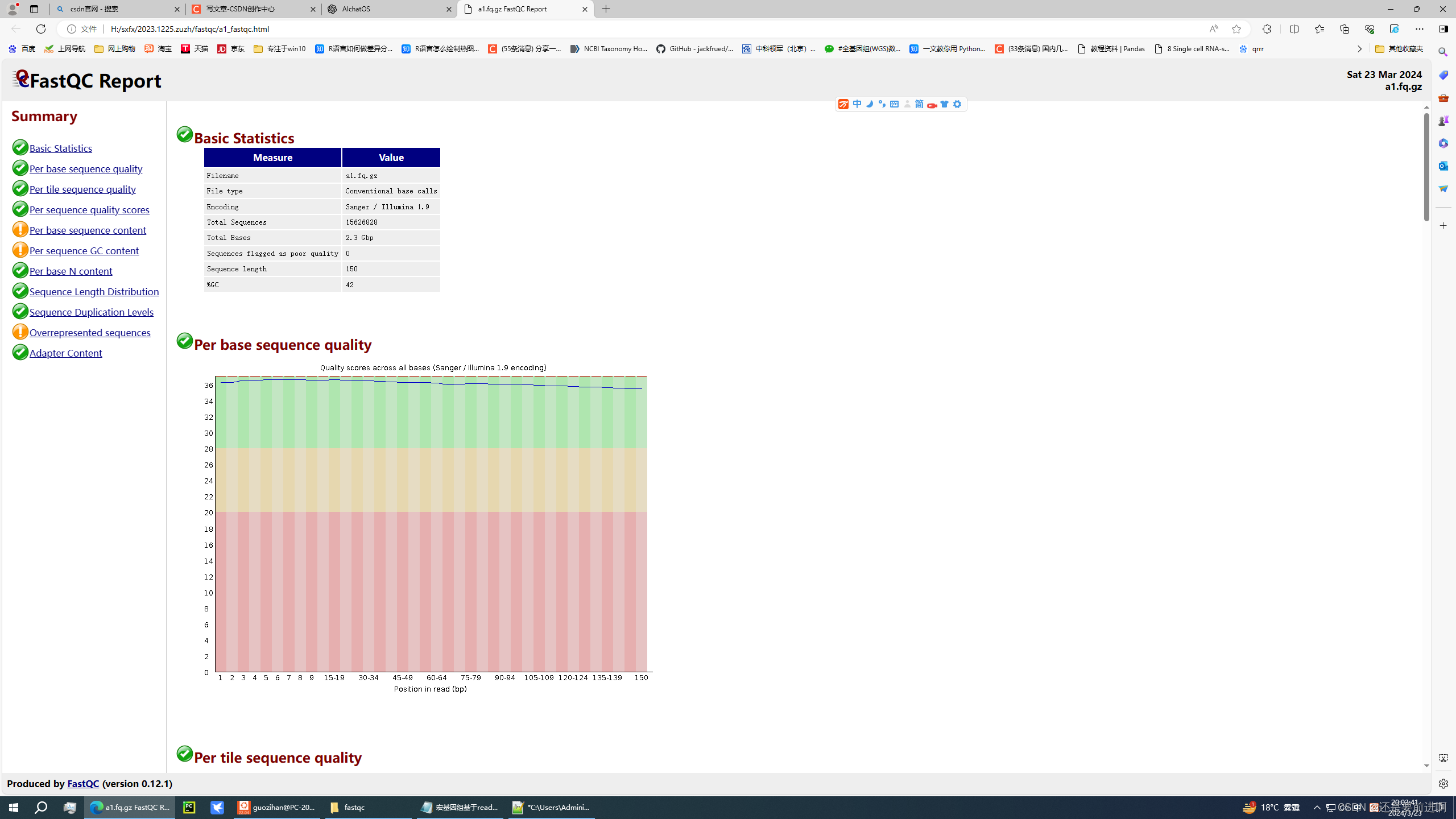
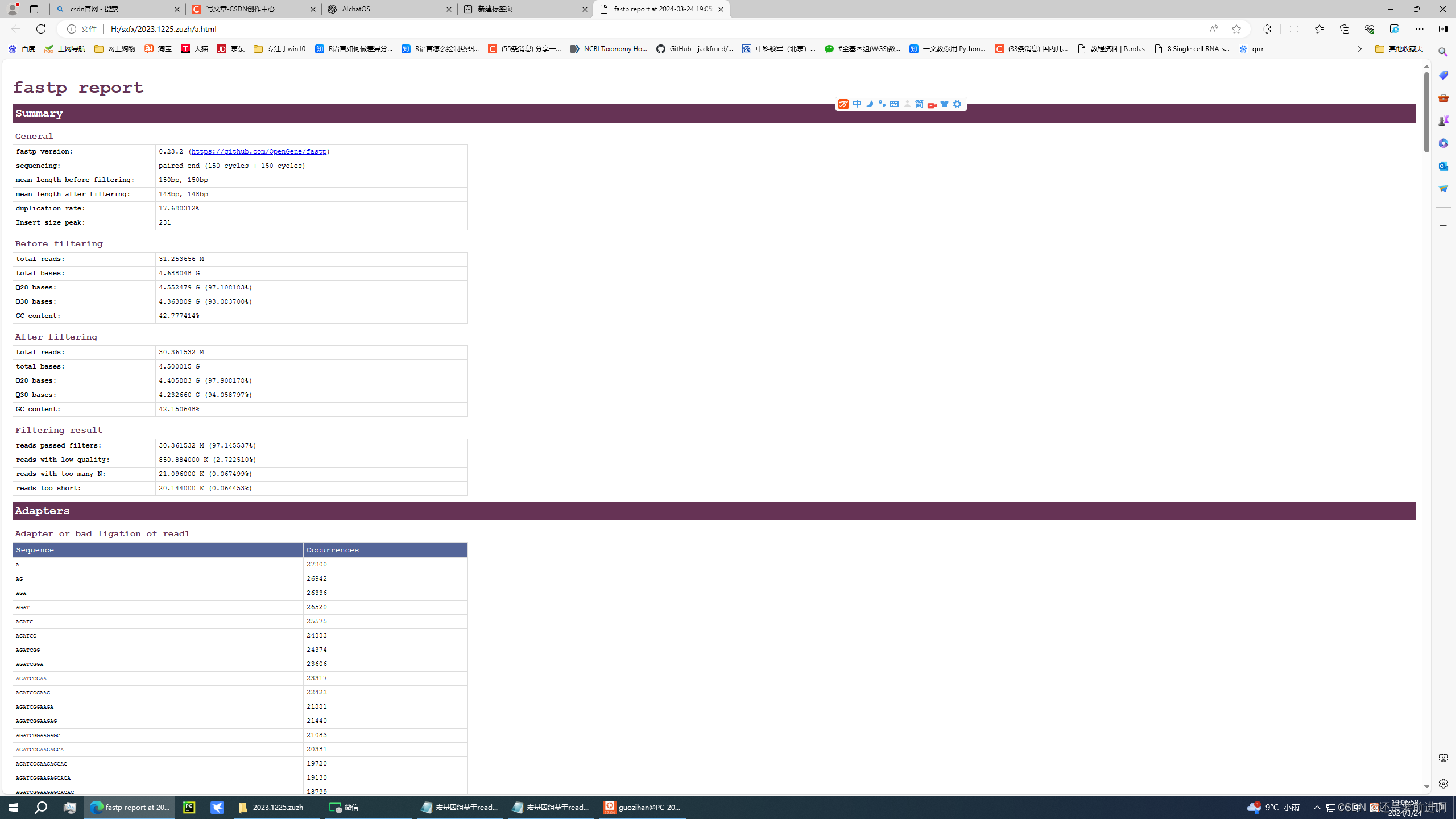
1. 质量控制
首先,需要对原始的测序数据(FASTQ格式)进行质量控制。通常使用FastQC进行初步的质量检查,然后使用Trimmomatic或者其他工具进行质量修剪。
# 质量检查
fastqc *.fq.gz
# 质量修剪
for sample in TH1 TH2 TH3 TW1 TW2 TW3
do
trimmomatic PE ${sample}.R1.fq.gz ${sample}.R2.fq.gz \
${sample}.R1.trim.fq.gz ${sample}.R1.untrim.fq.gz \
${sample}.R2.trim.fq.gz ${sample}.R2.untrim.fq.gz \
SLIDINGWINDOW:4:20 MINLEN:25
done
2.读段比对
使用比对工具(如HISAT2, STAR等)将质量修剪后的读段比对到参考基因组。
- 使用HISAT2进行读段比对
# 假设参考基因组索引为genome_index
for sample in TH1 TH2 TH3 TW1 TW2 TW3
do
hisat2 -x genome_index -1 ${sample}.R1.trim.fq.gz -2 ${sample}.R2.trim.fq.gz \
-S ${sample}.sam
done
- 使用STAR进行读段比对
- 下载并构建STAR索引
在使用STAR进行比对之前,需要首先构建或下载参考基因组的索引。这是一个一次性的过程。假设你已经有了参考基因组的fasta文件和基因注释文件(GTF格式),使用以下命令构建索引:
STAR --runThreadN NumberOfThreads \
--runMode genomeGenerate \
--genomeDir /path/to/genomeDir \
--genomeFastaFiles /path/to/genome/fasta \
--sjdbGTFfile /path/to/annotations.gtf \
--sjdbOverhang ReadLength-1
其中,NumberOfThreads是使用的线程数,/path/to/genomeDir是存储索引的目录,/path/to/genome/fasta和/path/to/annotations.gtf分别是参考基因组的fasta文件和基因注释文件的路径。ReadLength-1是读段长度减去1,这是一个重要的参数,需要根据数据来设定。
2. 读段比对
for sample in TH1 TH2 TH3 TW1 TW2 TW3
do
STAR --genomeDir /path/to/genomeDir \
--readFilesIn ${sample}.R1.trim.fq.gz ${sample}.R2.trim.fq.gz \
--runThreadN 10 \
--outFileNamePrefix ${sample} \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate
done
在这里,/path/to/genomeDir是你的索引目录,NumberOfThreads是使用的线程数, s a m p l e . R 1. t r i m . f q . g z 和 {sample}.R1.trim.fq.gz和 sample.R1.trim.fq.gz和{sample}.R2.trim.fq.gz分别是质量修剪后的读段文件。–outSAMtype BAM SortedByCoordinate会输出排序后的BAM文件,这对后续分析非常有用。
HISAT2和STAR的区别如下:
设计和算法:
HISAT2:使用了一种称为分层索引的方法。它首先将参考基因组划分为较小的片段,并为这些片段构建全局和局部的索引。这种方法使得HISAT2在处理基因组比对时特别有效,尤其是在考虑剪接位点和变异位点时。
STAR:使用了一种称为基于稀疏后缀数组的方法。STAR的这种方法允许它在一个单独的步骤中完成比对和剪接位点的检测,这使得它在处理大型数据集时非常快速和内存高效。性能和资源消耗:
HISAT2:通常比STAR使用更少的内存,但在处理时间上可能稍长。它特别适合于资源受限的计算环境。
STAR:虽然需要较多的内存,但在执行速度上通常比HISAT2更快,特别是在处理包含大量剪接事件的复杂转录组时。准确性和灵敏度:
HISAT2:在处理较长的读段和考虑基因组变异(如SNPs和小的indels)时表现出较高的准确性。
STAR:在检测剪接事件和转录本多样性方面表现优异,尤其是在处理短读段数据时。适用性: HISAT2:由于其对于基因组变异的考虑,HISAT2在进行种群遗传学研究或者需要高精度比对的场景中表现更好。 STAR:在进行转录组学研究,尤其是需要快速处理大规模数据集和复杂剪接事件的情况下更为适用。
易用性和功能: 两者都具备易用性和丰富的功能集,包括对多线程的支持、与常用转录组分析流程的兼容性等。
综上,选择HISAT2还是STAR取决于你的具体需求,比如数据的类型、可用资源、分析的焦点等。在某些情况下,研究者可能会根据不同阶段的需求选择不同的工具。例如,在初步筛选或快速分析时使用STAR,而在需要更细致分析基因组变异或长读段数据时使用HISAT2。
3.标记和去除PCR重复
使用Picard或samtools来标记和去除重复的读段。
对于使用samtools,流程如下:
for sample in TH1 TH2 TH3 TW1 TW2 TW3
do
samtools view -bS ${sample}.sam | \
samtools sort -o ${sample}.sorted.bam
samtools markdup ${sample}.sorted.bam ${sample}.dedup.bam
done
对于使用Picard, 流程如下:
for sample in TH1 TH2 TH3 TW1 TW2 TW3
do
samtools view -bS ${sample}.sam | \
samtools sort -o ${sample}.sorted.bam
picard MarkDuplicates I=${sample}.sorted.bam \
O=${sample}.dedup.bam M=${sample}.marked_dup_metrics.txt
done
4.转录组组装和表达量估计
使用工具如StringTie或Cufflinks对SAM/BAM文件进行转录组组装,生成转录本。
for sample in TH1 TH2 TH3 TW1 TW2 TW3
do
stringtie -e -B -G reference.gtf -o ${sample}.gtf ${sample}.dedup.bam
done
-e 选项告诉StringTie只估计已知转录本(在提供的GTF文件中)的表达量。
-B 选项会生成每个基因和转录本的表达量计数文件,这些文件可用于后续差异表达分析。
-G reference.gtf 是参考基因组的注释文件,这里需要你提供正确的路径和文件名。
5.差异表达分析
最后,使用DESeq2或edgeR等工具进行差异表达分析,以识别实验组和对照组之间差异表达的基因。
library(DESeq2)
# 假设countData为一个矩阵,行为基因,列为样本,包含基因的计数
# colData为DataFrame,包含样本信息
# 这里需要根据实际情况准备countData和colData
dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData,
design = ~ condition)
dds <- DESeq(dds)
res <- results(dds)
# 保存结果
write.csv(as.data.frame(res), file="DESeq2_results.csv")