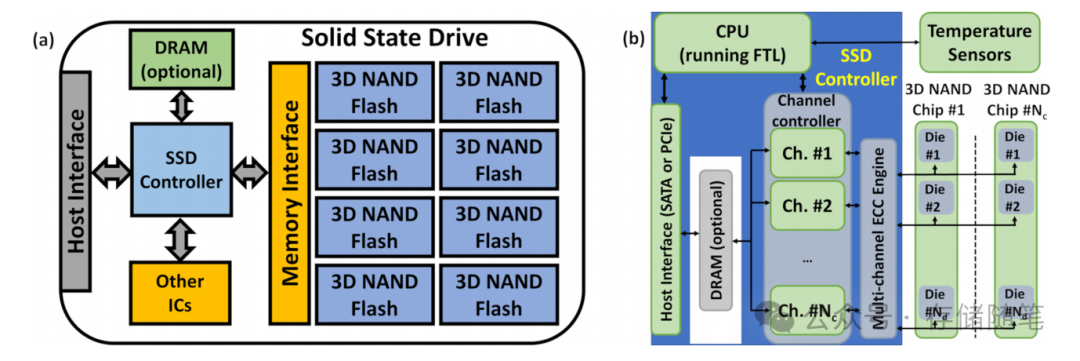
转载自:MetaAI
在生物学研究中,随着实验和计算技术的进步,生物系统研究产生了大量高通量数据。技术努力主要集中在提高吞吐量、降低成本和提升实验与计算效率。因此,整合不同类型组学数据,并通过关联分析识别关键因素和机制的计算方法变得尤为重要。

发表在《Nature Protocols》中的这篇文章,提出了一个可以从多组学中推断因果关系的系统框架 - Transkingdom Network Analysis (TkNA),并详细介绍了该框架的使用流程。TkNA是一种独特的因果推理分析框架,能够整合多个数据集和不同类型的组学数据,执行荟萃分析并识别关键的调控关系。它之前被用于研究抗生素耐药微生物、2型糖尿病和免疫缺陷相关肠病,以及宫颈癌、淋巴瘤和黑色素瘤中微生物组的作用。TkNA可以识别微生物和微生物基因、宿主途径、宿主基因以及控制宿主-微生物群相互作用的主要调节因素。
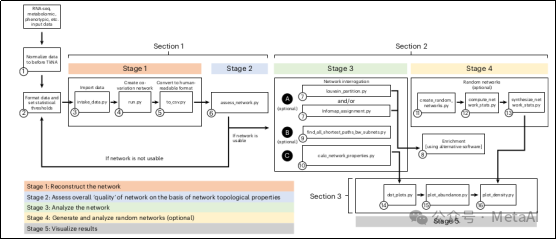
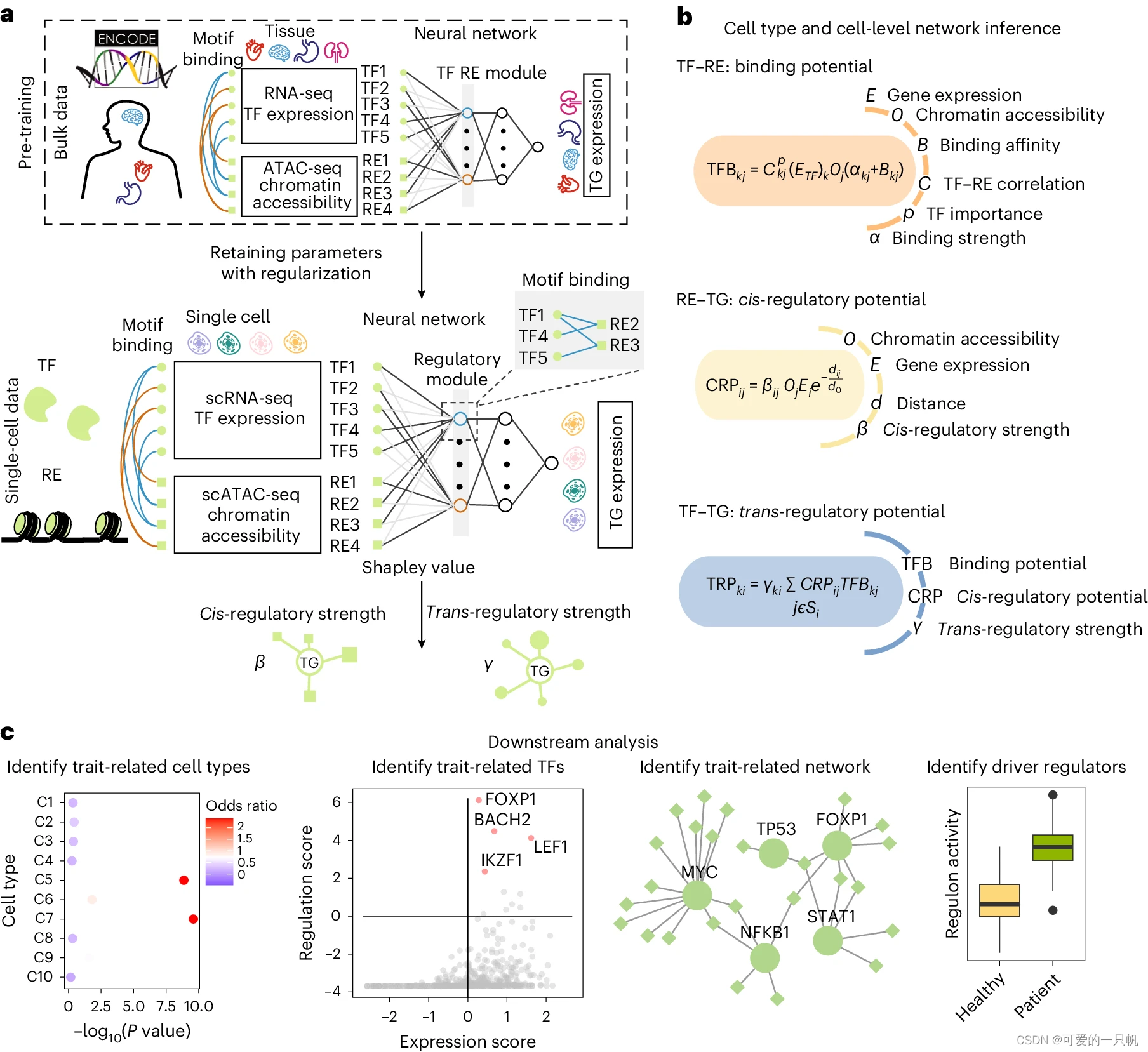
图1. TkNA流程图
TkNA流程包括3个主要部分,5个阶段,并在这些之前有两个预处理步骤(图1)。TkNA可以用于分析实验验证后的结果,构成一个循环的分析框架(图2)。
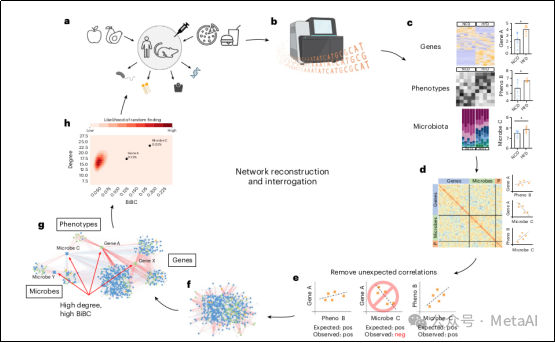
图2. TkNA管道以循环方式进行
a、进行实验并获得样品。b,在开始TkNA之前对样品进行测序和标准化。c、对每种数据类型进行比较。bar图分别代表测量的基因表达水平、微生物丰度和表型。星号代表治疗组之间的显着差异,因为仅保留显着变化的特征下游分析。d、在每种数据类型之间和内部执行相关性。e,删除意外的相关性。f、重建网络。g,询问网络以寻找调控节点。h,确定从网络中找到前部节点的概率,然后进行后续验证研究并重复该循环。
TkNA不仅可以识别关键调节节点,还能计算网络的拓扑属性,并通过如Cytoscape这样的外部程序可视化网络。TkNA的方法已经用于验证多项研究中的推论。它提供了一种分析不同组学数据之间交互作用的方法,可以用于分析遗传和转录数据、代谢物、蛋白质和表型之间的相互作用。与其他方法相比,TkNA侧重于通过荟萃分析识别跨多个队列的稳健模式,并且专注于建立因果关系而非仅仅发现关联。尽管TkNA提供了强大的工具,但用户仍需要具备一定的统计知识来理解软件的适用性和局限性。
TkNA的目标用户是宿主和/或微生物组领域的研究人员,这些人员可能缺乏计算和统计专业知识。该方法适用于生物和生物医学研究的多个学科,从建立新的细胞和分子治疗靶点到研究基础生物学问题。用户无需编程专业知识,但需要熟悉Unix环境中的命令行操作,以及能够理解JSON文件格式以自定义和修改程序选项。
该手册详细描述了一个复杂的分析流程和具体步骤,并给出了相应的命令行,这个流程分为3个主要部分,涵盖了5个阶段,以及在这些阶段之前的两个预处理步骤(图1)。下面只做简单描述,具体详细步骤见原文:
第一部分:重建网络
这部分涉及数据的标准化、文件格式化以及设定统计阈值(预处理步骤)。
阶段1:数据导入、计算/荟萃分析和按用户指定的条件过滤数据。这一步骤首先找出基于用户定义的统计标准的不同类别样本(如病态与健康对照组)间表达/丰富的变量(基因、微生物、代谢物等)。然后,进行每组内和组间的相关性分析。
阶段2:基于网络的拓扑特性,用户决定是否进入下一个分析阶段。这些特性包括网络密度、观察到的正/负相关偏差及意外相关比例等。
第二部分:询问/分析重建的网络
阶段3:分析重建的网络以找出在调查的生物过程中因果作用的节点或节点组。用户可以使用TkNA识别网络中的节点集群,并通过外部推荐软件进行富集分析,以识别集群中的节点所贡献的生物途径或功能。
第三部分:从用户重建的网络分析中创建发布就绪的图表
阶段4:评估特定节点显示非随机值的概率。在这里,TkNA重建了许多随机网络,与重建网络进行比较。
阶段5:创建多种高质量的图表,包括度分布的点图、节点及其计算属性的点图,以及前调节节点的丰富度或表达水平。
图3展示了TkNA的一些输出结果。
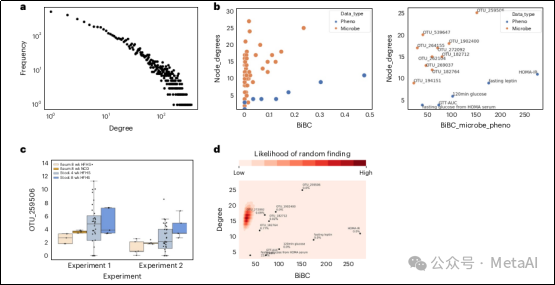
图3. TkNA生成的示例图
a,网络属性度(degree)分布图。b,左:节点属性可视化示例,其中每个点代表重建网络中的一个节点。右图:同一张图,放大了前10个微生物BiBC节点。c,b中前部BiBC节点的丰度/表达图示例。图例显示了数据集中的两个类。在本例中,将名为“高”的样本类别与名为“低”的样本类别进行比较。盒子显示每次实验每组的四分位数;须线包括除边远点之外的其余分布。d,10,000个随机网络的二维密度图。
TkNA方法依赖于在多个实验中进行荟萃分析,以识别多个队列中倍数变化(fold change)和相关性的稳健模式。默认情况下,它使用Fisher方法来组合来自多个独立测试的P值。其他通用R包(例如,meta、netmeta和mixmeta)提供了多种元分析方法,但这些方法并未考虑因果关系原理,例如相关不等式。其他R软件包(例如MixOmics、MOFA+和iClusterPlus)也使用复杂的统计方法来组合从同一患者测量的多个组学数据。然而,它们同时应用于多个队列或独特的组学数据(其中数据组成或不满足分布假设)可能具有挑战性。TkNA提供了一个框架来实现多种组学类型和群组的同时整合。请注意,“整合”一词指的是两种截然不同的分析。具体来说,在整篇文章中以以下方式使用它:荟萃分析是来自多个独立数据集的数据的集成,而网络重建涉及在多种类型的组学数据之间建立统计依赖关系的集成。
参考文献:
Newman, N.K., Macovsky, M.S., Rodrigues, R.R. et al. Transkingdom Network Analysis (TkNA): a systems framework for inferring causal factors underlying host–microbiota and other multi-omic interactions. Nat Protoc (2024). https://doi.org/10.1038/s41596-024-00960-w