Zhang Wei, Wu Chenjun, Xing Feiyang, Jiang Mingfeng, Zhang Yixuan, Liu Qi, Shi Zhuoxing, Dai Qi, scHybridBERT: integrating gene regulation and cell graph for spatiotemporal dynamics in single-cell clustering, Briefings in Bioinformatics, Volume 25, Issue 2, March 2024, bbae018, scHybridBERT: integrating gene regulation and cell graph for spatiotemporal dynamics in single-cell clustering | Briefings in Bioinformatics | Oxford Academic
论文地址:
论文代码:
摘要
图学习模型在单细胞RNA测序(scRNA-seq)数据的计算分析中受到了越来越多的关注。与传统的深度神经网络相比,图神经网络和语言模型通过从原始基因计数矩阵中提取图结构数据表现出了更优越的性能。现有的基于深度神经网络的聚类方法通常侧重于时间表达模式,而忽略了基因层面和细胞层面的内在交互,这可以被视为单细胞数据中的空间动态。在多视图建模框架下,基因–基因和细胞–细胞的交互能够提高细胞类型检测的性能。在本研究中,提取了时空嵌入和细胞图,以捕捉分子层面的空间动态。为了提高细胞类型检测的准确性,本研究提出了scHybridBERT架构,利用提取的时空模式对scRNA-seq数据进行多视图建模。在该scHybridBERT方法中,图学习模型用于处理细胞图,而Performer模型则利用时空嵌入。基准scRNA-seq数据集的实验结果表明,所提出的scHybridBERT方法通过整合时空嵌入和细胞图,能够提高单细胞聚类任务的准确性。
引言
单细胞RNA测序(scRNA-seq)技术为探索细胞异质性和高分辨率识别细胞亚群提供了强有力的工具。与传统的群体RNA测序相比,scRNA-seq数据允许在单个细胞水平上分析基因表达,从而更准确地呈现不同细胞类型和亚群的转录组特征。尤其是,scRNA-seq数据聚类被认为是识别和表征这些亚群的关键步骤,因为它能够检测可能对应于不同细胞类型或状态的独特转录组特征。由于细胞类型检测对于选择目标分子和通路至关重要,深度神经网络广泛应用于下游任务,如细胞类型注释和细胞聚类[1, 2]。
开发用于scRNA-seq数据的深度学习方法已经被用来揭示驱动复杂生物过程的分子和调控机制,包括疾病进展和细胞分化。对于人类来说,衰老过程被广泛认为与细胞类型分布和基因表达模式的变化有关。利用scRNA-seq数据建模可以识别衰老过程中的关键因素并发现潜在的调控机制。具体的疾病,包括神经退行性疾病,被认为与单细胞转录组密切相关[3, 4]。为了获得具有生物学意义的模式,构建了诸如AMP-AD和AMP-PD数据库等多组学数据平台,用于识别生物标志物并研究与神经退行性疾病相关的分子机制。为了处理异质数据集,多模态学习方法和特征融合技术使得从遗传学和转录组学中识别分子生物标志物和检测疾病亚型成为可能[5-9]。
然而,由于高水平的生物噪声和掉落事件(dropout events),导致假零计数,scRNA-seq数据的准确聚类仍然面临挑战。对于单细胞聚类任务,已经提出了许多基于深度学习的方法来从基因表达数据中检测细胞亚群[10-12]。网络结构和学习目标的设计可能会考虑单细胞数据的特征,旨在下游任务中实现高精度和可靠性[13, 14]。这些聚类方法涉及特征选择和降维,以减轻掉落事件和其他生物噪声对单细胞数据的影响[15-17]。由于信号模式挖掘能力强,复杂的深度学习模型包括生成模型和transformers也被应用于单细胞数据的分析和建模。然而,现有的深度学习模型通常专注于单细胞RNA测序数据的时间模式,而在一定程度上忽略了空间模式。从基因表达数据中提取的图结构数据被视为典型的时间模式。
深度学习模型由于其在各种下游任务中展示的卓越能力,包括细胞类型注释以及网络推断,被广泛应用于分析单细胞数据[18]。在这些深度学习模型中,scDeepCluster算法利用深度嵌入聚类,通过显式建模scRNA-seq数据的生成来学习特征表示[19]。作为另一种细胞聚类方法,SIMLR为基因表达矩阵学习适当的多核权重。该SIMLR方法从输入的单细胞数据中获得一个适当的细胞间相似性度量,并构建一个对称的相似性矩阵。无监督的DESC方法也利用深度嵌入[20],但配备了一个迭代自学习范式,旨在克服批次效应的挑战。不仅限于深度神经网络,大型语言模型在单细胞转录组学的计算分析中也发挥了作用。自然语言处理的快速进展由双向编码器表示(BERT)模型推动[21]。大型语言模型在单细胞数据分析中的一个应用是单细胞BERT模型[22],它利用基因表达数据揭示细胞的转录模式。
现有基于深度学习的单细胞数据分析方法主要关注单一视角,如标志基因的异常表达模式。这种做法通常忽略了细胞或基因之间的内在功能相互作用。这些有向相互作用以图结构数据的形式存在,可能有助于提高下游任务的准确性,包括单细胞聚类[23]。在先前的研究中,拓扑特征被视为RNA-seq数据的一种深层特征[24, 25]。为了处理图结构特征,各种基于图神经网络的方法被开发出来,以捕捉细胞和基因之间的相互作用[26-29]。此外,图神经网络在处理图结构特征方面表现出卓越的性能。一个可能的解释是,图结构特征提供了潜在的细胞-细胞和基因-基因相互作用,以提高模型在下游任务中的性能。细胞-细胞相似性或相互作用在下游任务中被视为有价值的信息。对于scRNA-seq数据,多模态学习成为集成时间模式和图结构特征的一个有前途的解决方案。
本研究提出了一种基于多视角建模的scHybridBERT框架,通过时空嵌入和细胞图识别细胞类型。该多视角建模结构包括分子水平的空间和时间动态。为了进行多视角建模,细胞图和空间嵌入捕捉调控系统的拓扑特征,而基因和表达嵌入被视为时间模式。为了提取关于基因-基因相互作用的动态,该scHybridBERT架构通过从scRNA-seq数据推断基因共表达网络来计算空间嵌入。同时,使用一种新颖的指数曼哈顿(Exp-Mah)相似性度量构建细胞图,并由图注意力网络(GATs)处理。随后,应用一种基于自适应多层感知机(MLP)的融合策略来集成包括时空嵌入和图结构数据在内的混合数据模态。对多个具有细胞类型标签的scRNA-seq数据集进行实验,以说明scHybridBERT方法的可行性和有效性。
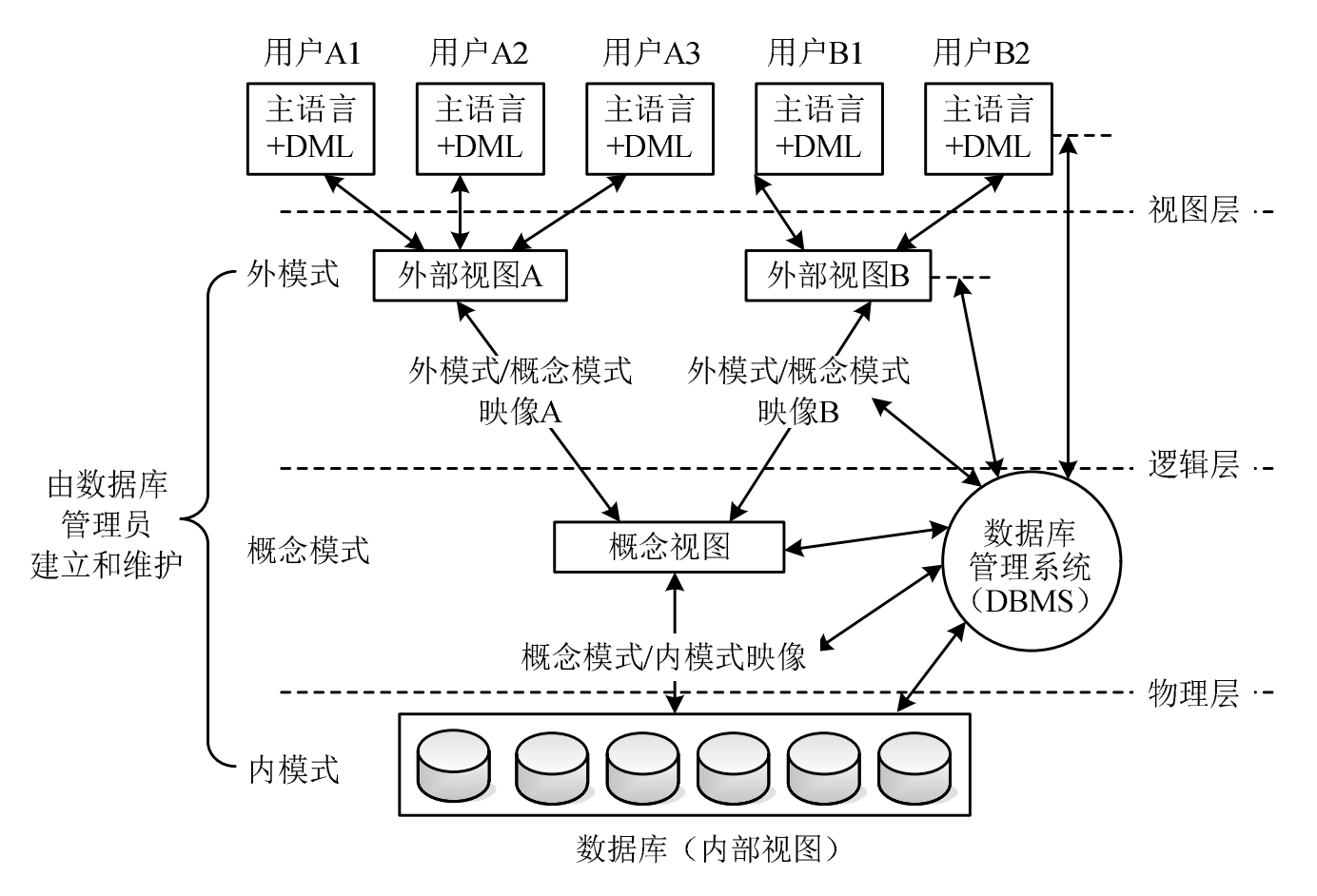
BERT模型的背景
现有研究表明,BERT架构在单细胞RNA测序(scRNA-seq)数据的计算分析中表现出色。在BERT架构中,transformers模型能够检查整个数据集的依赖关系,从而学习全局上下文。通过在无标签的scRNA-seq数据上进行自监督式的掩码训练,transformers模型及其衍生模型探索整个数据集的依赖关系,从而捕获全局序列并检测细微的依赖关系。预训练和微调机制在传统的scBERT模型中发挥了重要作用。
原始scRNA-seq数据的预处理
由于原始数据可能包含生物噪声和技术伪影,对原始数据矩阵进行特征工程是必不可少的。通常,原始计数矩阵中的非零表达值通常仅占约10%。因此,基因表达值被转换以确保正态性。同时,保留所有基因的表达,以获得关于基因图和细胞图的全面视角,以供后续分析使用。
由于在BERT架构中已经考虑了token嵌入和位置嵌入,因此在分析scRNA-seq数据时,有必要充分利用基因序列的特性。token嵌入是离散变量,而原始表达输入是代表具有生物噪声的基因表达的连续变量。采用词袋技术将基因表达进行分箱,这可以视为细胞中基因转录频率。
对于scRNA-seq数据,基因嵌入和表达嵌入被计算为预训练语言模型的输入。scBERT采用的预训练机制只能分析预训练期间学到的遗传特征。如果给定scRNA-seq数据集中没有包含在预训练参数中的基因,它们会被直接移除,从而影响模型的准确性和泛化性能。在这种情况下,scHybridBERT提出了一种端到端框架,具有很强的泛化性能。
基因嵌入
基因嵌入是通过gene2vec算法获得的,用于表示基因身份,并作为相对嵌入来捕捉共表达方面的语义相似性。共表达的基因保留更接近的表示形式,基因的分布式表示有助于捕捉基因间的关系。
这一部分借鉴了word2vec的思想,学习基因表达的连续向量表示,使基因序列中的每个片段映射到连续向量空间中的一个向量表示。这些向量具有语义意义,使相似的基因片段在向量空间中更加接近。gene2vec算法的目标函数定义如下:
\[ L = -\sum_{w \in D} \sum_{c \in C_w} \log P(c|w) \]
其中,D是包含所有基因的文本语料库,C_w是与基因w相关的上下文集合。gene2vec算法采用skip-gram机制来学习词向量。给定一个基因w,目标是最大化与该基因相关的上下文基因c的条件概率P(c|w)。具体来说,这个条件概率可以通过以下公式计算:
\[ P(c|w) = \frac{e^{v_c \cdot v_w}}{\sum_{c' \in D} e^{v_{c'} \cdot v_w}} \]
其中,v_w和v_c分别是基因w和c的向量表示。具体来说,gene2vec算法用于生成基因嵌入,以捕捉基因之间的语义相似性。这些基因嵌入与离散化的表达嵌入一起,扮演了Performer模型的时间嵌入的角色,从而在分子水平上提取时间信息。基因嵌入表示来自gene2vec的基因身份,属于第一个分箱,而表达嵌入与基因表达相关,属于第二个分箱,并按照与基因嵌入相同的方向进行转换。
时间表达嵌入
除了基因嵌入,每个基因的转录水平作为连续变量提供了有价值的信息。基因嵌入和表达嵌入之间的区别在于它们对scRNA-seq数据的不同视角。类似于语言模型,基因表达水平被认为是生物系统中的类似事件。因此,采用词频分析方法将连续表达变量进行分箱离散化,从而获得200维的向量作为模型训练的标记嵌入。
基因嵌入指的是将基因序列信息转换为向量表示的方法,其主要目的是描述基因之间的相似性和差异性。而表达嵌入不同,它是一种将基因表达谱数据转换为向量表示的方法,旨在描述基因在不同条件下的表达情况。
Performer模型
Performer模型可以用来克服数据的高维度和噪声,并揭示单细胞转录组数据的底层图结构。作为引入自注意机制的深度神经网络模型,Performer模型具有降低空间复杂度的注意机制的优势。Performer被视为Transformer的扩展,基于自注意机制。这种机制具有在有限空间复杂度下实现类似注意机制效果的优势,并且在处理序列数据时以较少的计算资源高效地捕捉长距离依赖关系。这使得Performer在处理大规模单细胞测序数据时具有更好的性能和可扩展性。在Performer中,自注意机制表示如下公式:
\[ \text{Attention}(Q, K, V) = D^{-1}(QK^T)V, \quad D = \text{diag}(QK^T1_L), \]
其中,Q是查询向量,K和V分别表示键向量和值向量,\(d_k\)表示查询向量的维度。该公式可以用于计算每个细胞与其他细胞之间的相似性,从而将它们分组为不同的细胞子集。通过使用低秩近似和随机特征技术,Performer将自注意机制的计算复杂度从\(O(n^2)\)降低到\(O(n \log n)\),其中n是序列的长度。这表明Performer提高了计算注意矩阵的效率,并展示了比标准Transformer模型更强的处理长序列的能力。
scHybridBERT框架概述
受到语言模型的启发,提出了scHybridBERT方法来对scRNA-seq数据进行多视角建模,从而获得更全面的模型。Performer模型和图神经网络分别利用时空动态和细胞图等异质信息。随后,通过基于自适应多层感知器(MLP)的融合策略将时空嵌入和图结构数据进行整合。Performer模型旨在处理长单细胞RNA序列,捕捉全局信息,而图神经网络则发现复杂的聚类结构并补偿孤立样本点的不足。scHybridBERT方法的基本示意图如图1所示。
在该示意图中,分子系统的全局拓扑信息由基因调控网络和共表达网络包含。这种拓扑信息尚未明确纳入以往的单细胞聚类方法中。通过逐元素相加将时空嵌入结合起来,同时应用结合了曼哈顿距离和相关系数的Exp-Mah距离度量来学习关于细胞级相互作用的深层特征。在该多视角建模结构的另一个管道中,变分自编码器计算的潜在特征被用于训练图注意网络(GATs)。随后,通过基于自适应MLP的融合策略整合异质数据模态。
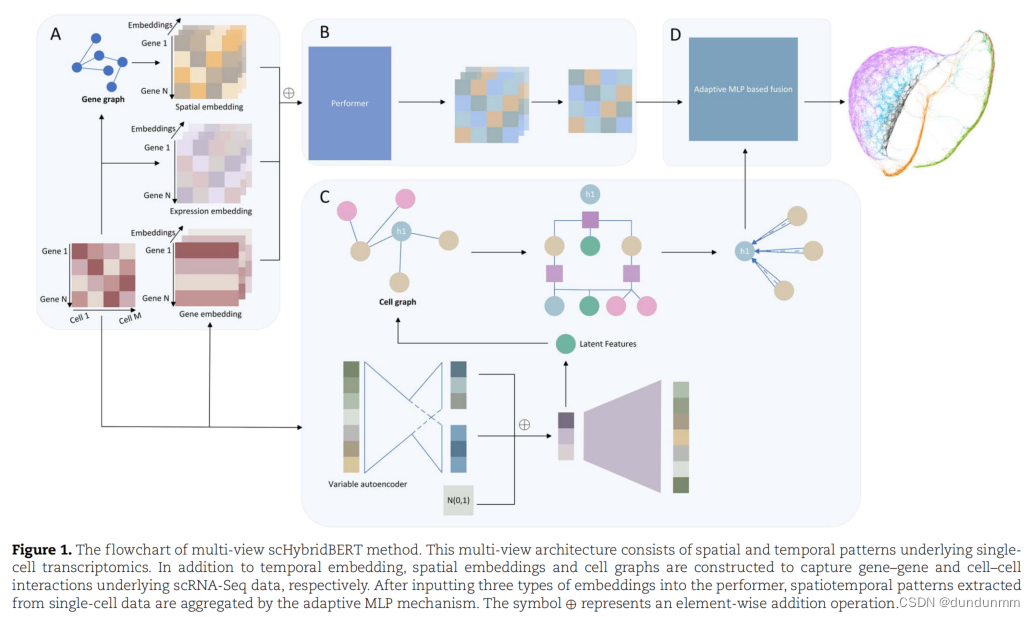
时空表达嵌入的构建
从scRNA-Seq数据中提取的时空嵌入中,基因共表达网络被计算为空间嵌入,以捕捉分子水平上的基因-基因相互作用。所提出的scHybridBERT方法从单细胞RNA测序数据中推断基因共表达网络,并利用邻域信息获得基因-基因相互作用。提取的空间嵌入为关于RNA-seq数据的计算建模提供了拓扑视角。基因间的共表达关系定义如下:
\[ \cos(\theta) = \frac{\mathbf{x} \cdot \mathbf{y}}{\|\mathbf{x}\| \|\mathbf{y}\|} = \frac{\sum_{i=1}^{n} x_i y_i}{\sqrt{\sum_{i=1}^{n} x_i^2} \sqrt{\sum_{i=1}^{n} y_i^2}}, \]
其中,\(\mathbf{x}\) 和 \(\mathbf{y}\) 表示两个基因的向量表达,\(x_i\) 和 \(y_i\) 分别表示向量中第 \(i\) 个元素的值,\(n\) 表示特征向量的维度。在这项工作中,使用node2vec算法捕捉基因-基因关系以嵌入模式。给定当前顶点 \(v\),访问下一个顶点 \(x\) 的概率描述公式如下:
\[ P(c_i = x \mid c_{i-1} = v) = \begin{cases}
\frac{\pi_{vx}}{Z}, & \text{if} \ (v, x) \in E, \\
0, & \text{otherwise}
\end{cases} \]
其中,\(\pi_{vx}\) 是顶点 \(v\) 和 \(x\) 之间的未归一化转移概率,\(Z\) 是归一化常数。Node2vec引入了两个超参数 \(p\) 和 \(q\) 来控制随机游走的策略,假设当前随机游走经过边 \((t, v)\) 并到达顶点 \(v\)。设 \(\pi_{vx} = \alpha_{pq} \cdot w_{vx}\),其中 \(w_{vx}\) 表示顶点 \(v\) 和 \(x\) 之间的边权重,\(\alpha_{pq}\) 的定义如下:
\[ \alpha(t, x) = \begin{cases}
\frac{1}{p}, & \text{if} \ d_{tx} = 0, \\
1, & \text{if} \ d_{tx} = 1, \\
\frac{1}{q}, & \text{if} \ d_{tx} = 2
\end{cases} \]
其中,\(d_{tx}\) 表示顶点 \(t\) 和 \(x\) 之间的最短路径距离。作为参数,变量 \(p\) 控制重复访问刚访问过的顶点的概率。如果 \(p\) 较高,则减少访问刚访问过的顶点的概率。参数 \(q\) 控制随机游走是向外还是向内。如果 \(q > 1\),则随机游走倾向于访问靠近 \(t\) 的顶点,反之,则倾向于访问远离 \(t\) 的顶点。作为一种节点嵌入算法,增加 \(p\) 并减少 \(q\) 可以捕捉基因间的共表达信息,同时提高算法效率。
细胞图的构建
单细胞RNA测序技术可以从单个细胞中生成大量的基因表达数据。在这种技术中,原始基因计数矩阵通常具有高维度,而细胞数量相对有限,使得直接分析表达矩阵并揭示细胞关系变得困难。为了解决这个问题,使用VAE模型来降低基因表达矩阵的维度,并获得低维特征。这些与表达相关的特征被表示为编码细胞的嵌入。在嵌入表达之后,scHybridBERT框架进一步构建细胞图以捕捉细胞间的相互作用,目的是在细胞聚类任务中整合空间动态。
VAE模型的编码器部分包含两个全连接层,fc1和fc2,以及一个ReLU激活函数。VAE模型的编码器定义如下:
\[ \mu = \text{fc21}(\text{relu}(\text{fc1}(x))) \]
\[ \log \sigma^2 = \text{fc22}(\text{relu}(\text{fc1}(x))), \]
其中,\(\mu\) 和 \(\log \sigma^2\) 分别表示后验均值和对数方差。使用参数化技巧得到潜变量 \(z\) 如下:
\[ z = \mu + \sigma \epsilon, \ \epsilon \sim N(0, I). \]
VAE模型的解码器部分由两个全连接层组成,分别为fc3和fc4,以及一个sigmoid激活函数。该部分用于将潜变量 \(z\) 解码回原始输入 \(x\) 的重建,如下:
\[ h3 = \text{relu}(\text{fc3}(z)) \]
\[ x' = \text{sigmoid}(\text{fc4}(h3)). \]
VAE模型的编码器块将输入数据映射到潜在空间,解码器块从潜在表示中重建输入。VAE模型的重建损失计算如下:
\[ L_{MSE} = \frac{1}{N} \sum_{i=1}^{N} \| x_i - f(g(x_i)) \|^2, \]
其中,\( f(g(x_i)) \) 表示AE模型的输出结果,\( x_i \) 是基因表达值的序列。 \| x_i - f(g(x_i)) \|^2 表示序列的2-范数,可以进一步解释为 \(\sum_{j=1}^{M} |\delta_j| \),其中 \(\delta_j\) 是某个细胞的第 \(j\) 个值,即其基因的表达量。
该VAE模型通过重建损失函数进行训练,包括均方误差(MSE)和平均绝对误差。仅仅使用重建损失可能无法保证生成的潜在表示的连续性和可解释性。为了控制潜在表示的分布,使用Kullback–Leibler (KL)散度作为正则化项来约束潜在表示的分布。在本研究中,KL散度用于测量学习到的潜在表示与先验分布之间的差异,定义如下:
\[ L_{KL} = \frac{1}{2} \sum_{i=1}^{d} (1 + \log(\sigma_i^2) - \mu_i^2 - \sigma_i^2), \]
其中,\(\mu\) 和 \(\sigma\) 分别表示由自动编码器获得的潜在表示的均值和标准差,\(d\) 表示潜在表示的维度。该损失函数的目标是将潜在表示的分布约束为标准正态分布。最终的损失函数包括两个部分,表示如下:
\[ L = \alpha L_{MSE} + (1 - \alpha) L_{KL}, \]
其中 \( L_{MSE} \) 表示重建损失函数,\(\alpha\) 和 \(\beta\) 表示正则化参数,用于平衡两个损失函数的权重。经过训练VAE模型后,生成了对应于细胞的基因表达的低维表示。因此,提取了细胞之间的相似性和差异,并显著降低了计算复杂性。
Exp-Mah度量捕捉细胞-细胞相互作用
在先前的研究中,使用欧氏距离度量来衡量两个样本之间的功能相似性,通过计算它们之间的欧氏距离。然而,欧氏距离在高维数据中受到维度诅咒的影响。这表明欧氏距离对异常值非常敏感,而在基因表达矩阵中常见到异常值。为了缓解这个问题,scHybrid-BERT架构采用了新颖的指数曼哈顿(Exp-Mah)距离,全面考虑基因表达水平的相关值。
这里的曼哈顿距离 dM(x, y) 定义如下:
\[ dM(x, y) = \sum_{i=1}^{N} |x_i - y_i| \]
其中, x 和 y 分别代表两个不同细胞的n维基因向量,xi 和 yi 表示两个细胞上第i个基因的值,N 表示细胞中的基因数。
使用Exp-Mah距离时,将两个向量投影到高斯平面上并计算距离。在降维之后,基因序列的特征变得独立并且包含正负值。相关系数可以全面考虑正负特征,计算如下:
\[ Rcor(x, y) = \frac{\sum_{i=1}^{n}(x_i - \bar{x})(y_i - \bar{y})}{\sqrt{\sum_{i=1}^{n}(x_i - \bar{x})^2 \sum_{i=1}^{n}(y_i - \bar{y})^2}} \]
其中, x 和 y 是来自两个不同细胞的n维基因向量,xi 和 yi 分别表示两个细胞上第i个基因的值,¯x 和 ¯y 是两个向量的均值,N 表示细胞中的基因数。
因此,衡量细胞相似性 Sim(x, y) 的公式定义如下:
\[ \text{sim}(x, y) = Rcore^{\alpha dM} \]
其中,α 是调整两个测量指标权重的超参数。指数函数在独立变量增加时增长迅速。直接线性组合可能导致相似性分数范围不一致。Exp-Mah距离中的指数函数可以结合两者,统一相似性分数的范围。
构建细胞图使用K最近邻(KNN)算法,其中每个节点代表一个单细胞,节点之间的边表示细胞之间的相似性或关联。构建细胞图需要设置邻居的数量K,这与捕捉到的细胞相互作用图的规模相关。每个细胞节点在K个最短距离内找到其邻居细胞,并在它们之间创建边。因此,K的值影响构建的细胞图的密度和复杂度。较大的K值会导致细胞图中更密集的边。构建细胞图揭示了单细胞RNA-seq数据中重要的细胞相互作用模式。
细胞图的修剪
图学习方法作为提议的scHybridBERT模型的核心部分,并利用细胞图来检测细胞亚群。图神经网络已被用于大型图上的节点级表示学习。图神经网络的关键组成部分之一是聚合过程,它将节点邻居的信息组合起来更新其表示。图学习中的聚合过程可以分解为三个主要步骤:采样、消息传递和聚合。在本研究中,图神经网络被用作聚合方法,聚合函数定义如下:
\[ h_{\text{agg}}^{(i)} = \text{ReLU} \left( \sum_{j \in N(i)} \frac{1}{c_{ij}} W h_{\text{in}}^{(j)} \right) \quad \text{(16)} \]
这里,\( h_{\text{in}}^{(i)} \) 表示节点的输入表示,\( h_{\text{agg}}^{(i)} \) 表示聚合后的表示,\( W \) 是可学习的权重矩阵,\( N(i) \) 表示节点 \( i \) 的邻居节点集合,\( c_{ij} \) 是由以下公式定义的归一化因子:

其中,\( d_i \) 和 \( d_j \) 分别是节点 \( i \) 和 \( j \) 的度数。聚合函数被视为节点邻居输入表示的归一化加权和。归一化因子 \( c_{ij} \) 考虑了节点的不同度数,并确保聚合过程不偏向具有更高度数的节点。聚合表示上应用了ReLU激活函数,逐元素地引入非线性。
使用细胞图训练GAT
与传统的图神经网络相比,GAT在处理大规模图,特别是涉及数千个节点的细胞图时,具有增强的能力。引入了注意力机制来学习邻居节点的权重,并获得节点本身的表达。这种机制能自动分配邻居节点的权重并捕捉节点之间的关系。在这种情况下,GAT模型能够捕捉图中节点之间的关系,实现对图结构数据的端到端学习。
图结构数据被表示为图 \( G = (V, E) \),其中 \( V \) 是节点集合, \( E \) 是边集合。通常,使用邻接矩阵 \( A \in \mathbb{R}^{N \times N} \) 来表示图的拓扑结构,其中 \( N \) 是节点数,\( A_{ij} \) 表示节点 \( i \) 和节点 \( j \) 之间是否有边。此外,我们还可以有特征矩阵 \( X \in \mathbb{R}^{N \times F} \),其中 \( F \) 是每个节点的特征数。
GAT的基本构建模块是图注意力层,输入特征矩阵 \( X \) 被线性转换成更新后的特征矩阵 \( H \in \mathbb{R}^{N \times F'} \),其中 \( F' \) 是输出特征的数量。这可以通过与可学习权重矩阵 \( W \in \mathbb{R}^{F \times F'} \) 的矩阵乘法实现,表示为式 (18):
\[ H = XW \quad \text{(18)} \]
这里,\( W \) 是一个可学习的权重矩阵。计算节点 \( i \) 和节点 \( j \) 之间的关联分数,以确定节点之间的注意力权重。这可以通过异质特征的内积和应用非线性激活函数来完成。在GAT模型中,这种非线性LeakyReLU激活函数的形式如下 (19):
\[ e_{ij} = \text{LeakyReLU} \left( \mathbf{a}^T [H_i \| H_j] \right), \quad \text{(19)} \]
这里,\(\|\) 表示向量级联操作,\( \mathbf{a} \in \mathbb{R}^{2F'} \) 是一个可学习的权重向量,\( H_i \) 和 \( H_j \) 分别表示节点 \( i \) 和节点 \( j \) 的特征向量。为了学习节点 \( i \) 相对于其邻居节点的注意力权重,使用softmax函数对节点的关联分数进行归一化。归一化后的注意力权重 \( \alpha_{ij} \) 按照式 (20) 计算:
\[ \alpha_{ij} = \frac{\exp(e_{ij})}{\sum_{k \in N(i)} \exp(e_{ik})}, \quad \text{(20)} \]
这里,\( N(i) \) 表示节点 \( i \) 的邻居。为了更新节点 \( i \) 的特征向量,使用注意力权重计算邻居节点特征的加权和,表示为式 (21):
\[ H'_i = \sum_{j \in N(i)} \alpha_{ij} H_j, \quad \text{(21)} \]
其中,\( H'_i \) 是节点 \( i \) 的更新后的特征向量。为了增强模型的预测能力,GAT模型采用了多头注意力机制,其中包含多个独立的注意力机制。每个注意力头的输出特征向量被串联或平均以获得最终输出。多头注意力层的输出可以表示为式 (22):
\[ H' = \frac{1}{K} \sum_{k=1}^{K} \text{GAT}_k(H), \quad \text{(22)} \]
这里,\( \text{GAT}_k \) 表示第 \( k \) 个注意力头,\( K \) 是注意力头的数量。通过堆叠多个图注意力层,GAT模型能够学习细胞图的复杂表示。引入注意力机制使GAT能够有效捕捉节点之间的关系,并进行端到端学习。
scHybridBERT方法的伪代码描述如算法1。
在算法1中,第5步旨在计算与图1中潜在变量相对应的h(i),而第9步计算elvj是使用细胞图训练GATs的过程。

实验
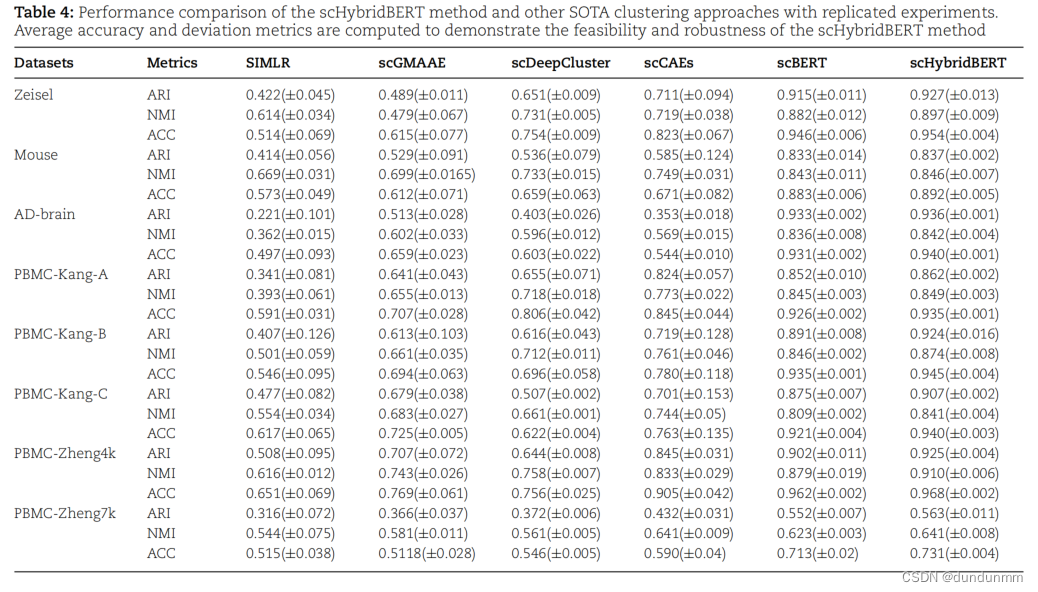
的基础上,进行了多视图数据的融合分析。



![[<span style='color:red;'>论文</span><span style='color:red;'>阅读</span>]DETR](https://img-blog.csdnimg.cn/direct/e8df1ed8695749d1b661ff6ad6833838.png)
![[<span style='color:red;'>论文</span><span style='color:red;'>阅读</span>]BEVFusion](https://img-blog.csdnimg.cn/direct/65697d2df3dc47d2a67baf3c2e53c330.png)











![[代码复现]Self-Attentive Sequential Recommendation](https://img-blog.csdnimg.cn/direct/ce09b3d9ac0f4d58a2d794a7e2fd1694.png)