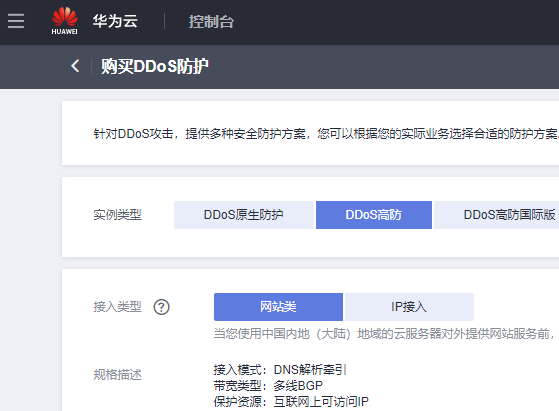
# 生态学研究人员通常对微生物群落的功能特征感兴趣,因为功能或代谢数据对于解释微生物群落的结构和动态以及推断其潜在机制是强有力的。
# 由于宏基因组测序复杂且昂贵,利用扩增子测序数据预测功能谱是一个很好的选择。
# 有几个软件经常用于此目标,如PICRUSt, Tax4Fun和FAPROTAX。
# 这些工具可以很好地用于基于测序结果的原核生物群落的功能谱预测。此外,获得每个分类群或OTU的功能也很重要,
# 而不仅仅是整个群落的概况。但是很难知道每个OTU的确切功能。FAPROTAX数据库是根据已发表在书籍和文献中的已知研究成果,
# 对原核生物的性状和特征进行的汇总。我们将原核生物的分类信息与该数据库进行比对,以确定原核生物在生物地球化学作用上的特征。
# 我们还实现了FUNGuild和FungalTraits数据库来识别真菌性状。
> t1 <- trans_func$new(dataset = dataset)
> t1$cal_spe_func(prok_database = "FAPROTAX")
> t1$cal_spe_func(fungi_database = "FungalTraits")
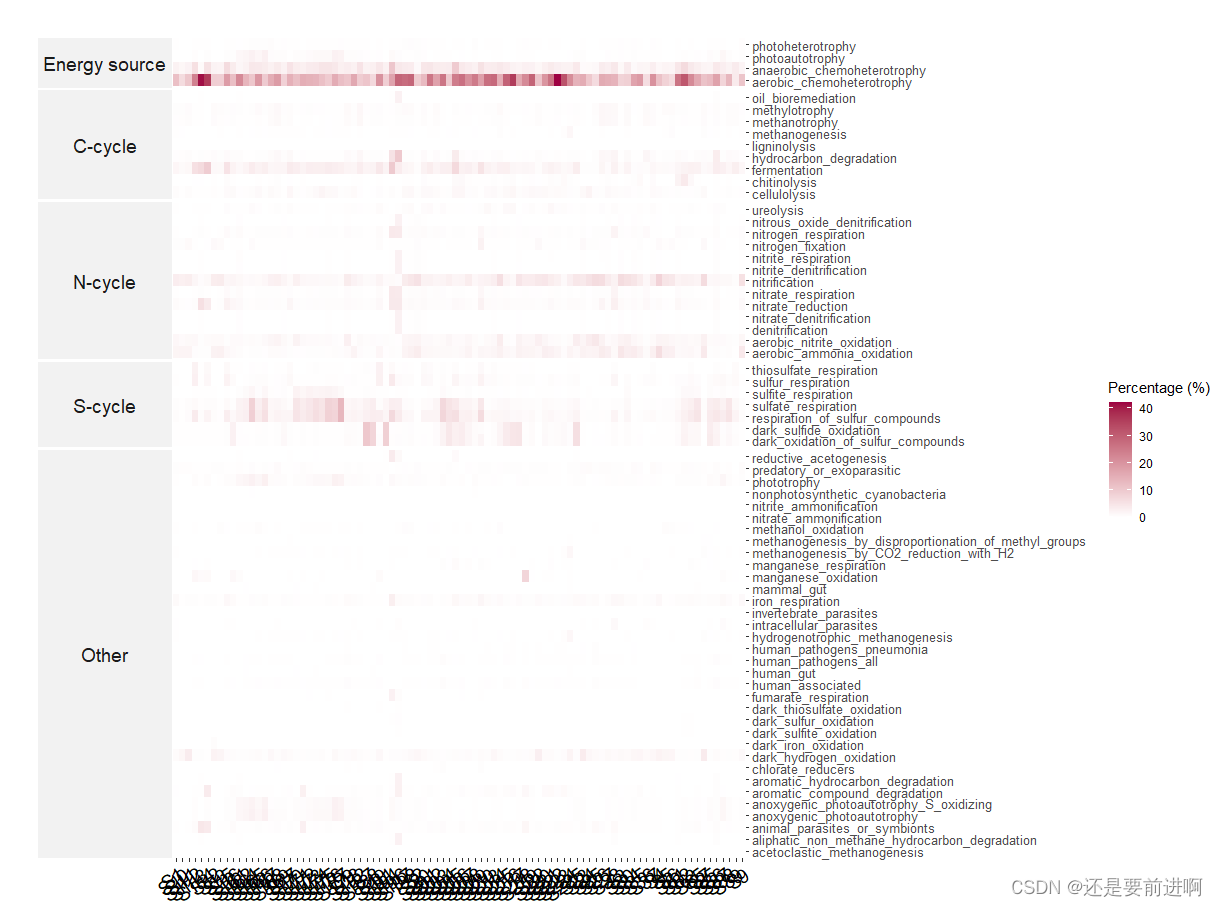
# 计算群落中具有特定性状的物种百分比。具有特定性状的分类群所占百分比可以反映群落中相应的功能潜力。
# 因此,该方法是一种不考虑类群间系统发育距离的功能冗余表示。
> t1$cal_spe_func_perc(abundance_weighted = TRUE)
> t1$show_prok_func(use_func = "methanotrophy")
> t1$plot_spe_func_perc()
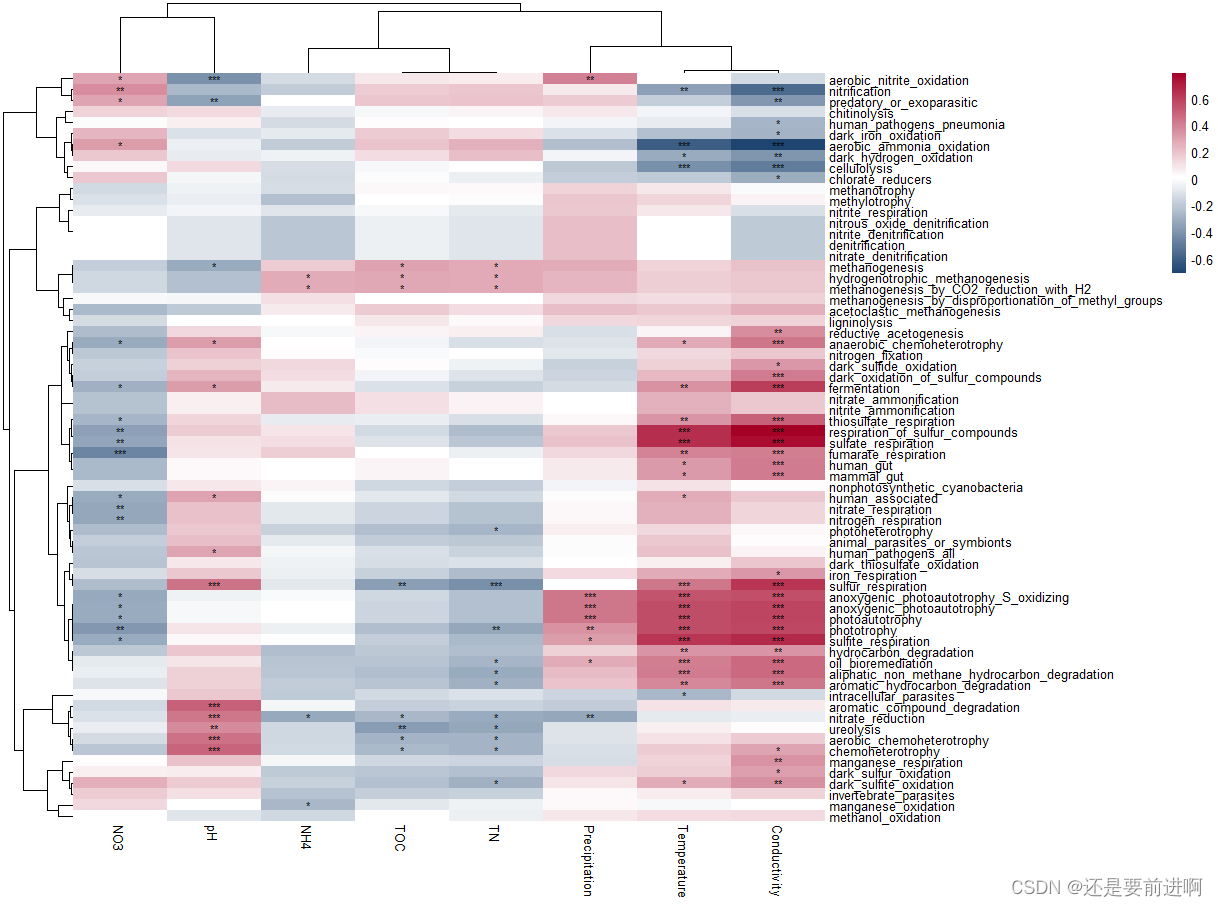
#然后我们尝试将社区的res_spe_func_perc与环境变量联系起来
> t3 <- trans_env$new(dataset = dataset, add_data = env_data_16S[, 4:11])
> t3$cal_cor(add_abund_table = t1$res_spe_func_perc, cor_method = "spearman")
> library(pheatmap)
> t3$plot_cor(pheatmap = TRUE)
microeco就先分享到这里,这个包比较复杂,我只是分享了部分,想要学习,得从事具体的项目。这些代码,我都跑过一边,大家可以先跑跑。