大家好,有人在生信技能树群里问:如何使用seurat v5读取v4创建的文件
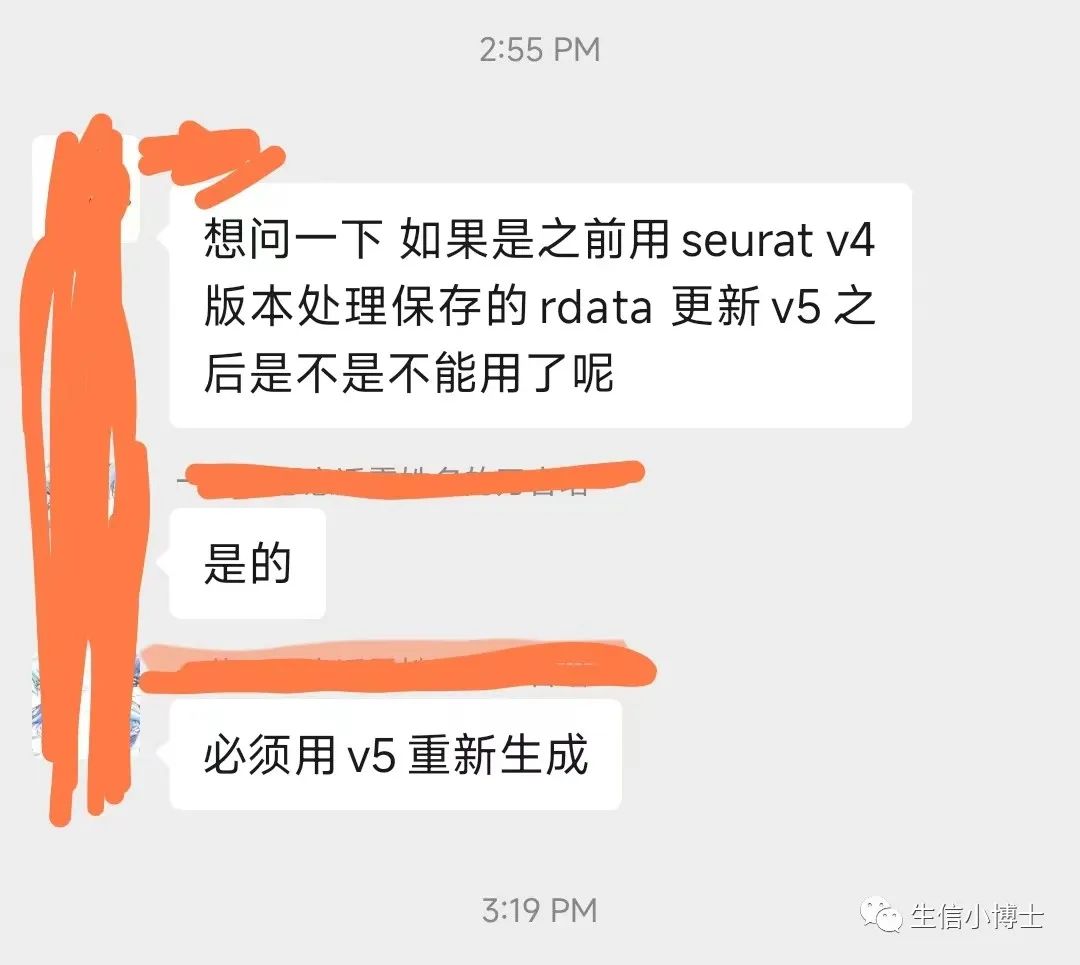
我直觉告诉我不要重新生成,肯定有简便方法。
一 Seurat官网都有哪些内容
我们去官网文档来找个简便方法:https://satijalab.org/seurat/

好像没有说seurat v5和v4对象可以直接转换,别着急,咱继续看
二 Seuratv5和v4的区别到底在哪里
官网上都说啦:https://satijalab.org/seurat/articles/announcements.html

主要有这5点区别:
1 Seurat 对象和 Assay 类发生了变化:
支持更多种类的检测和数据类型,包括磁盘上的矩阵。
引入了分层结构来存储数据,例如原始计数、标准化数据和 z 得分/方差稳定数据。
可以使用
$访问器或LayerData函数来访问数据。现有的 Seurat v4 函数和工作流程仍可在 v5 中使用。
2 整合工作流程发生了变化:
简化了整合流程,提高了速度和内存效率。
整合结果与 v4 工作流程略有不同,但用户仍可在 v5 中运行 v4 整合工作流程。
所有数据可以保存为单个对象,整合前可以拆分层。
3 差异表达分析发生了变化:
使用 presto 包(如果可用)进行差异表达分析,以加快速度。
在计算 log-FC 时,在组级别而不是细胞级别实施伪计数,这可能会导致更高的 logFC 估计值,但也可能更不稳定。
4 SCTransform v2 发生了变化:
默认使用 SCTransform 的更新版本 2。
用户可以设置
vst.flavor = "v1"参数来运行以前的工作流程。
5 Pseudobulk analysis发生了变化(伪批量分析):
鼓励使用
AggregateExpression函数进行伪批量分析,以降低噪音、提高低表达基因的定量并减少数据矩阵的大小。
但是还是没看到seurat v5和v4对象可以直接转换,别着急,咱继续看
三 万能的google与github给出了答案
#https://github.com/satijalab/seurat/issues/7409# convert a v5 assay to a v3 assaypbmc3k[["RNA3"]] <- as(object = pbmc3k[["RNA"]], Class = "Assay")# convert a v3 assay to a v5 assaypbmc3k[["RNA5"]] <- as(object = pbmc3k[["RNA3"]], Class = "Assay5")
可以看到转换前后的大小变化
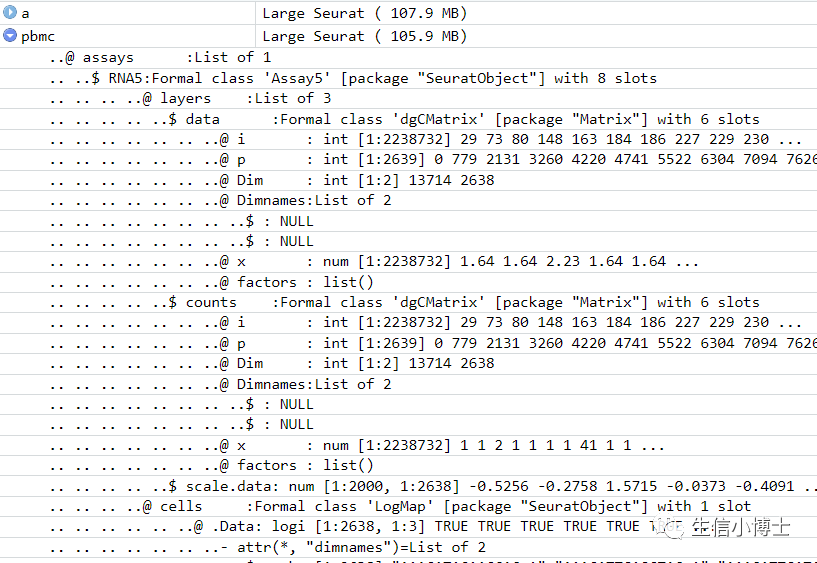
v3和v5都可以互相转换,应该不需要我写v4 和v5互相转换了吧

如果实在想看,你看今天的第二篇推文就行啦
后记
本文技术不难,更多的是一种思想方法,帮助大家去开拓自己的思路。
交流更多,加我微信,备注 姓名 学校


![]()
看完记得顺手点个“在看”哦!




















![[机器人-2]:开源MIT Min cheetah机械狗设计(二):机械结构设计](https://img-blog.csdnimg.cn/img_convert/a049ab5962d9c072a6574f1e1c7d4e2e.webp?x-oss-process=image/format,png)