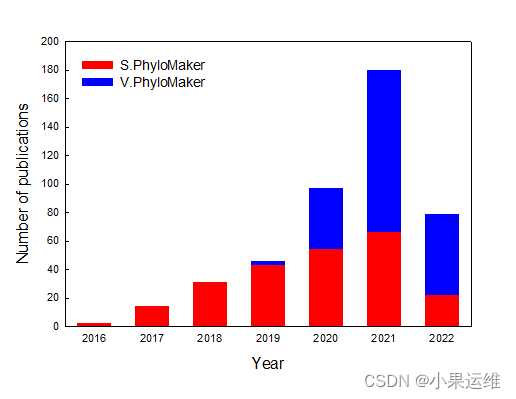
S.PhyloMaker介绍
S.PhyloMaker是一款用于生物系统学研究的软件工具,主要用于构建和分析种系发生树(也称为进化树)。以下是对S.PhyloMaker的详细介绍:
功能特性:
- 数据导入:S.PhyloMaker支持多种格式的分子序列数据导入,包括FASTA、PHYLIP、NEXUS等。
- 序列 alignment:软件内置了多种序列比对算法,可以帮助用户对输入的分子序列进行比对。
- 树构建:支持多种种系发生树构建方法,如最大似然法(Maximum Likelihood)、贝叶斯推理法(Bayesian Inference)、邻接法(Neighbor-Joining)和最大简约法(Minimum Evolution)等。
- 树优化:通过搜索最优树形结构和参数组合,优化种系发生树。
- 树解析:提供了一系列工具用于解析和可视化种系发生树,包括树的修剪、重新根定、分支长度调整、拓扑测试等。
- 分析功能:可以进行物种多样性分析、系统发育信号检测、选择压力分析等进化学分析。
用户界面:
- S.PhyloMaker的用户界面设计直观易用,提供了图形化的工作流程,使得用户可以轻松地完成从数据导入到结果分析的全过程。
系统要求:
- S.PhyloMaker是一款跨平台的软件,可以在Windows、Mac OS和Linux操作系统上运行。
- 对硬件的要求取决于数据集的大小和复杂性,一般来说,需要足够的内存和处理器性能来处理大规模的数据和复杂的计算任务。
应用领域:
- S.PhyloMaker在生物系统学、分子进化、微生物组学、群体遗传学等多个领域中有广泛的应用。
S.PhyloMaker使用步骤
S.PhyloMaker的使用步骤和一些常用命令的介绍:
安装S.PhyloMaker包:
install.packages("S.PhyloMaker")
加载S.PhyloMaker包:
library(S.PhyloMaker)创建物种分支(phylogenies):
a. 从NCBI进行下载: download.ncbi()
b. 使用自定义数据创建: create.phylo(file, format)
读取物种分支:
a. 从NCBI下载的phylo文件: read.ncbi(file)
b. 从指定文件中读取phylo文件: read.phylo(file, format)
转换物种分支的格式:
a. 转换至ape包的phylo对象格式: to.ape(file)
b. 转换至phylotools包的phylo4d对象格式: to.phylo4d(file)
物种分支的可视化:
a. 绘制物种分支树状图: plot.phylo(file)
b. 绘制分支颜色标签(branch color): plot.phylo(file, color)
物种分支的编辑和处理:
a. 删除物种分支: prune.phylo(file, species)
b. 裁剪物种分支的范围: crop.phylo(file, start, end)
c. 在物种分支上添加标签: label.phylo(file, labels)
d. 提取物种分支的信息: extract.phylo(file, column)
物种分支数据的导出:
a. 导出为NEXUS格式: export.nexus(file, output)
b. 导出为Newick格式: export.newick(file, output)
具体使用方法
S.PhyloMaker是一个用于处理系统发育数据的R工具包。它可以从原始数据中创建系统发育树,并进行一系列的数据处理和可视化操作。下面是使用步骤和常用命令的介绍:
步骤:
安装R:首先需要安装R语言环境,可以从R官方网站(https://www.r-project.org/)下载对应系统的安装包进行安装。
安装S.PhyloMaker:打开R控制台,在命令行中输入以下命令来安装S.PhyloMaker包:
install.packages("S.PhyloMaker")
- 加载S.PhyloMaker包:在R控制台中输入以下命令来加载S.PhyloMaker包:
library(S.PhyloMaker)
- 创建系统发育树:
- 使用原始数据创建系统发育树:
tree <- phylo_maker(data = mydata, type = "raw")
这里,mydata是原始数据,type参数指定数据类型为"raw"。
- 从已有的系统发育树文件创建系统发育树:
tree <- phylo_maker(data = "tree_file.nwk", type = "tree")
这里,tree_file.nwk是已有的系统发育树文件路径,type参数指定数据类型为"tree"。
- 数据处理和分析:
- 提取叶子节点和内部节点:
leaves <- get_leaves(tree)
nodes <- get_nodes(tree)
- 计算节点的度:
degree <- get_degree(tree)
- 计算节点的高度:
height <- get_height(tree)
- 计算节点的深度:
depth <- get_depth(tree)
- 进行系统发育相关的分析:
phylotools_analysis(tree)
- 数据可视化:
- 绘制系统发育树:
plot_tree(tree)
- 绘制节点度的分布图:
plot_degree(tree)
- 绘制节点高度的分布图:
plot_height(tree)
github使用示例
library("phytools") # load the "phytools" package.
example<-read.csv("example.splist.csv",header=T) # read in the example species list.
phylo<-read.tree("PhytoPhylo.tre") # read in the megaphylogeny.
nodes<-read.csv("nodes.csv",header=T) # read in the nodes information of the megaphylogeny.
result<-S.PhyloMaker(spList=example, tree=phylo, nodes=nodes) # run the function S.PhyloMaker.
str(result) # the structure of the ouput of S.PhyloMaker.
par(mfrow=c(1,3),mar=c(0,0,1,0)) # show the phylogenies of the three scenarios.
plot(result$Scenario.1,cex=1.1,main="Scenarion One")
plot(result$Scenario.2,cex=1.1,main="Scenarion Two")
plot(result$Scenario.3,cex=1.1,main="Scenarion Three")S.PhyloMaker完成分析脚本(有动手能力的可以改改):
S.PhyloMaker<-function (tree, spList, nodes, output.spList = T, scenarios = c("S1", "S2", "S3"))
{
options(scipen=999)
tree0 <- tree
spList[sapply(spList, is.factor)] <- lapply(spList[sapply(spList, is.factor)], as.character)
if (any(duplicated(spList$species)))
{
warning("Duplicated species detected and removed.")
print(spList$species[duplicated(spList$species)])
}
spList <- spList[!duplicated(spList$species), ]
spList.original <- spList
spList$species <- gsub(" ", "_", spList$species)
spList$species <- gsub("(^[[:alpha:]])", "\\U\\1", spList$species, perl = TRUE)
spList$genus <- gsub("(^[[:alpha:]])", "\\U\\1", spList$genus, perl = TRUE)
spList$family <- gsub("(^[[:alpha:]])", "\\U\\1", spList$family, perl = TRUE)
rnN <- data.frame(node.label = paste("N", 1:length(tree$node.label), sep = ""), oriN = tree$node.label, stringsAsFactors = FALSE)
nodes[,c("level","family","genus","rn","bn","taxa")]<-lapply(nodes[,c("level","family","genus","rn","bn","taxa")], as.character)
tree$node.label <- paste("N", 1:length(tree$node.label), sep = "")
kk<-c()
for (i in 1:length(tree$tip.label)) {
kk<-c(kk,substring(tree$tip.label[i],1,gregexpr("_",tree$tip.label[i])[[1]][1]-1))
}
m<-data.frame(num=1:length(kk),genus=kk,species=tree$tip.label)
m<-merge(m,nodes[,c("genus","family")])
mX <- m
m <- m[,c("genus","family")]
m <- m[!duplicated(m$genus),]
dimnames(m)[[2]][2] <- "family_in_PhytoPhylo"
m<-m[,c("genus","family_in_PhytoPhylo")]
m0 <- spList[!duplicated(spList$genus), c("genus","family")]
dimnames(m0)[[2]][2] <- "family_in_spList"
mm<-merge(m0, m)
g<-mm[which(is.na(match(paste(mm$genus,mm$family_in_spList,sep="_"),paste(mm$genus,mm$family_in_PhytoPhylo,sep="_")))),]
if (dim(g)[1]>0)
{
print("Taxonomic classification not consistent between spList and PhytoPhylo.")
print(g)
}
add.tip <- spList[which(is.na(match(spList$species, tree$tip.label))), ]
status <- rep("match(prune)", dim(spList)[1])
status[which(is.na(match(spList$species, tree$tip.label)))] <- "match(add)"
if (dim(add.tip)[1] == 0 & length(na.omit(match(spList$species, tree$tip.label))) == 0)
stop("Incorrect format of species list.")
if (length(setdiff(spList$species, tree0$tip.label)) == 0 & length(na.omit(match(spList$species, tree$tip.label))) > 0)
{
print("There is no species needs to be added, all the species are pruned from PhytoPhylo.")
splis <- spList.original
treeX <- drop.tip(tree0, setdiff(tree0$tip.label, splis$species))
splis$status <- "match(prune)"
phylo0 <- list(Scenario.1 = NULL, Scenario.2 = NULL, Scenario.3 = NULL, Species.list = splis)
if ("S1" %in% scenarios) {phylo0$Scenario.1 <- treeX}
if ("S2" %in% scenarios) {phylo0$Scenario.2 <- treeX}
if ("S3" %in% scenarios) {phylo0$Scenario.3 <- treeX}
phylo0[sapply(phylo0, is.null)] <- NULL
return(phylo0)
stop()
}
add.tip$sort <- ""
add.tip$sort[which(!is.na(match(add.tip$genus, nodes[nodes$level == "G", ]$genus)))] <- "G1"
add.tip$sort[which(is.na(match(add.tip$genus, nodes[nodes$level == "G", ]$genus)) & !is.na(match(add.tip$family, nodes[nodes$level == "F", ]$family)))] <- "F1"
add.tip$sort[add.tip$sort == "F1"][duplicated(add.tip[add.tip$sort == "F1", ]$genus)] <- "F2"
a <- which(add.tip$sort == "")
if (length(a) > 0)
{
print(paste("Note:", length(a), "taxa unmatch:",sep=" "))
print(add.tip$species[a])
status[match(add.tip$species[a], spList$species)] <- "unmatch"
}
spList.original$status <- status
if ("S1" %in% scenarios) {
t1 <- tree
rnN1<-rnN
nG <- nodes[nodes$level == "G", ]
nF <- nodes[nodes$level == "F", ]
data <- add.tip[add.tip$sort == "F1", ]
if (dim(data)[1] > 0) {
for (i in 1:dim(data)[1]) {
n <- match(data$family[i], nF$family)
g <- nF$gen.n[n]
s <- nF$sp.n[n]
if (g == 1 & s == 1) {
num <- grep(nF$taxa[n], t1$tip.label)
len <- t1$edge.length[match(num, t1$edge[, 2])]
t1 <- bind.tip(t1, tip.label = data$species[i], edge.length = len, where = num, position = len)
nF$gen.n[n] <- g + 1
nF$sp.n[n] <- s + 1
num <- grep(nF$taxa[n], t1$tip.label)
t1$node.label[match(t1$edge[match(num, t1$edge[, 2]), 1], unique(t1$edge[, 1]))] <- paste("NN", t1$Nnode + 1, sep = "")
rnN1$node.label[match(nF$bn[n], rnN1$node.label)]<- paste("NN", t1$Nnode + 1, sep = "")
nF$bn[n] <- paste("NN", t1$Nnode + 1, sep = "")
nF$bn.bl[n] <- len
}
else {
num <- unique(t1$edge[, 1])[match(nF$bn[n], t1$node.label)]
len <- nF$bn.bl[n]
t1 <- bind.tip(t1, tip.label = data$species[i], edge.length = len, where = num)
}
}
}
data <- add.tip[add.tip$sort != "F1", ]
if (dim(data)[1] > 0) {
for (i in 1:dim(data)[1]) {
n <- grep(paste(data$genus[i], "_", sep = ""), t1$tip.label)
if (length(n) == 1) {
num <- n
len <- t1$edge.length[match(num, t1$edge[, 2])]
t1 <- bind.tip(t1, tip.label = data$species[i], edge.length = len, where = num, position = len)
}
if (length(n) > 1) {
num <- fastMRCA(t1, t1$tip.label[min(n)], t1$tip.label[max(n)])
len <- fastDist(t1, t1$tip.label[min(n)], t1$tip.label[max(n)])/2
t1 <- bind.tip(t1, tip.label = data$species[i], edge.length = len, where = num)
}
}
}
toDrop <- setdiff(1:length(t1$tip.label), which(!is.na(match(t1$tip.label, spList$species))))
t1 <- drop.tip(t1, tip = toDrop)
Re <- which(!is.na(match(t1$node.label, rnN1$node.label)))
noRe <- which(is.na(match(t1$node.label, rnN1$node.label)))
t1$node.label[Re] <- rnN1$oriN[match(t1$node.label, rnN1$node.label)[Re]]
t1$node.label[noRe] <- ""
}
else {
t1 <- NULL
}
if ("S2" %in% scenarios) {
t2 <- tree
rnN2<-rnN
nG <- nodes[nodes$level == "G", ]
nF <- nodes[nodes$level == "F", ]
data <- add.tip[add.tip$sort == "F1", ]
if (dim(data)[1] > 0) {
for (i in 1:dim(data)[1]) {
n <- match(data$family[i], nF$family)
g <- nF$gen.n[n]
s <- nF$sp.n[n]
if (g == 1 & s == 1) {
num <- grep(nF$taxa[n], t2$tip.label)
len <- t2$edge.length[match(num, t2$edge[,2])] * sample((1:99)/100,1)
t2 <- bind.tip(t2, tip.label = data$species[i], edge.length = len, where = num, position = len)
nF$gen.n[n] <- g + 1
nF$sp.n[n] <- s + 1
num <- grep(data$species[i], t2$tip.label)
t2$node.label[match(t2$edge[match(num, t2$edge[, 2]), 1], unique(t2$edge[,1]))] <- paste("NN", t2$Nnode + 1, sep = "")
rnN2$node.label[match(nF$bn[n], rnN2$node.label)]<- paste("NN", t2$Nnode + 1, sep = "")
nF$bn[n] <- paste("NN", t2$Nnode + 1, sep = "")
nF$bn.bl[n] <- len
}
else {
num <- unique(t2$edge[, 1])[match(nF$bn[n], t2$node.label)]
len <- t2$edge.length[match(num,t2$edge[,2])] * sample((1:99)/100,1)
t2 <- bind.tip(t2, tip.label = data$species[i], edge.length = len, where = num, position = len)
nF$gen.n[n] <- g + 1
nF$sp.n[n] <- s + 1
num <- grep(data$species[i], t2$tip.label)
t2$node.label[match(t2$edge[match(num, t2$edge[, 2]), 1], unique(t2$edge[, 1]))] <- paste("NN", t2$Nnode + 1, sep = "")
rnN2$node.label[match(nF$bn[n], rnN2$node.label)]<- paste("NN", t2$Nnode + 1, sep = "")
nF$bn[n] <- paste("NN", t2$Nnode + 1, sep = "")
nF$bn.bl[n] <- nF$bn.bl[n]+len
}
}
}
data <- add.tip[add.tip$sort != "F1", ]
if (dim(data)[1] > 0) {
for (i in 1:dim(data)[1]) {
n <- grep(paste(data$genus[i], "_", sep = ""), t2$tip.label)
if (length(n) == 1) {
num <- n
len <- t2$edge.length[match(num, t2$edge[, 2])] * sample((1:99)/100,1)
t2 <- bind.tip(t2, tip.label = data$species[i], edge.length = len, where = num, position = len)
}
if (length(n) > 1) {
num <- sample(n,1)
len <- t2$edge.length[match(num, t2$edge[, 2])] * sample((1:99)/100,1)
t2 <- bind.tip(t2, tip.label = data$species[i], edge.length = len, where = num, position = len)
}
}
}
toDrop <- setdiff(1:length(t2$tip.label), which(!is.na(match(t2$tip.label, spList$species))))
t2 <- drop.tip(t2, tip = toDrop)
Re <- which(!is.na(match(t2$node.label, rnN2$node.label)))
noRe <- which(is.na(match(t2$node.label, rnN2$node.label)))
t2$node.label[Re] <- rnN2$oriN[match(t2$node.label, rnN2$node.label)[Re]]
t2$node.label[noRe] <- ""
}
else {
t2 <- NULL
}
if ("S3" %in% scenarios) {
t3 <- tree
rnN3<-rnN
nG <- nodes[nodes$level == "G", ]
nF <- nodes[nodes$level == "F", ]
data <- add.tip[add.tip$sort == "F1", ]
if (dim(data)[1] > 0) {
for (i in 1:dim(data)[1]) {
n <- match(data$family[i], nF$family)
g <- nF$gen.n[n]
s <- nF$sp.n[n]
if (g == 1 & s == 1) {
num <- grep(nF$taxa[n], t3$tip.label)
len <- t3$edge.length[match(num, t3$edge[, 2])] * (2/3)
t3 <- bind.tip(t3, tip.label = data$species[i], edge.length = len, where = num, position = len)
nF$gen.n[n] <- g + 1
nF$sp.n[n] <- s + 1
num <- grep(nF$taxa[n], t3$tip.label)
t3$node.label[match(t3$edge[match(num, t3$edge[, 2]), 1], unique(t3$edge[, 1]))] <- paste("NN", t3$Nnode + 1, sep = "")
rnN3$node.label[match(nF$bn[n], rnN3$node.label)]<- paste("NN", t3$Nnode + 1, sep = "")
nF$bn[n] <- paste("NN", t3$Nnode + 1, sep = "")
nF$bn.bl[n] <- len
}
if (g == 1 & s > 1) {
num <- unique(t3$edge[, 1])[match(nF$bn[n], t3$node.label)]
if ((2/3)*nF$rn.bl[n] <= nF$bn.bl[n]) { len <-(nF$rn.bl[n]-nF$bn.bl[n])/2 }
if ((2/3)*nF$rn.bl[n] > nF$bn.bl[n]) { len <-nF$rn.bl[n]*2/3-nF$bn.bl[n] }
t3 <- bind.tip(t3, tip.label = data$species[i], edge.length = len, where = num, position = len)
nF$gen.n[n] <- g + 1
nF$sp.n[n] <- s + 1
num <- unique(t3$edge[, 1])[match(nF$bn[n], t3$node.label)]
t3$node.label[match(t3$edge[match(num, t3$edge[, 2]), 1], unique(t3$edge[, 1]))] <- paste("NN", t3$Nnode + 1, sep = "")
rnN3$node.label[match(nF$bn[n], rnN3$node.label)]<- paste("NN", t3$Nnode + 1, sep = "")
nF$bn[n] <- paste("NN", t3$Nnode + 1, sep = "")
nF$bn.bl[n] <- len+nF$bn.bl[n]
}
if (g > 1) {
num <- unique(t3$edge[, 1])[match(nF$bn[n], t3$node.label)]
len <- nF$bn.bl[n]
t3 <- bind.tip(t3, tip.label = data$species[i], edge.length = len, where = num)
}
}
}
data <- add.tip[add.tip$sort != "F1", ]
if (dim(data)[1] > 0) {
for (i in 1:dim(data)[1]) {
n <- grep(paste(data$genus[i], "_", sep = ""), t3$tip.label)
if (length(n) == 1) {
len <- t3$edge.length[match(n, t3$edge[, 2])]/2
t3 <- bind.tip(t3, tip.label = data$species[i], edge.length = len, where = n, position = len)
nG$sp.n[match(data$genus[i], nG$genus)] <- length(n) + 1
nu <- grep(paste(data$genus[i], "_", sep = ""), t3$tip.label)
num <- fastMRCA(t3, t3$tip.label[nu[1]], t3$tip.label[nu[2]])
t3$node.label[match(num, unique(t3$edge[, 1]))] <- paste("NN", t3$Nnode + 1, sep = "")
rnN3$node.label[match(nG$bn[n], rnN3$node.label)]<- paste("NN", t3$Nnode + 1, sep = "")
nG$bn[match(data$genus[i], nG$genus)] <- paste("NN", t3$Nnode + 1, sep = "")
nG$bn.bl[match(data$genus[i], nG$genus)] <- len
}
if (length(n) > 1) {
num <- fastMRCA(t3, t3$tip.label[min(n)], t3$tip.label[max(n)])
len <- as.numeric(branching.times(t3))[match(num, unique(t3$edge[, 1]))]
t3 <- bind.tip(t3, tip.label = data$species[i], edge.length = len, where = num)
}
}
}
toDrop <- setdiff(1:length(t3$tip.label), which(!is.na(match(t3$tip.label, spList$species))))
t3 <- drop.tip(t3, tip = toDrop)
Re <- which(!is.na(match(t3$node.label, rnN3$node.label)))
noRe <- which(is.na(match(t3$node.label, rnN3$node.label)))
t3$node.label[Re] <- rnN3$oriN[match(t3$node.label, rnN3$node.label)[Re]]
t3$node.label[noRe] <- ""
}
else {
t3 <- NULL
}
if (output.spList == FALSE)
spList <- NULL
phylo <- list(Scenario.1 = t1, Scenario.2 = t2, Scenario.3 = t3, Species.list = spList.original)
phylo[sapply(phylo, is.null)] <- NULL
return(phylo)
}相关阅读: