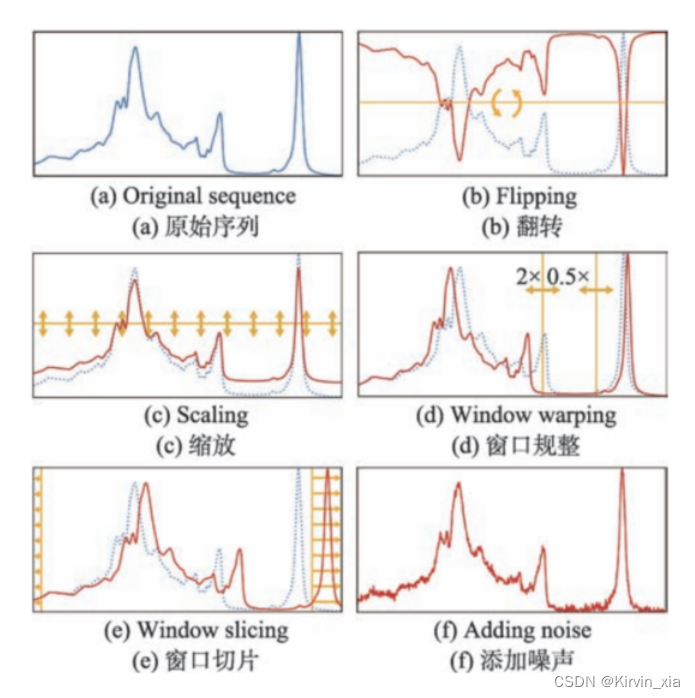
文献介绍

文献题目: The tidyomics ecosystem: enhancing omic data analyses
研究团队: Stefano Mangiola(澳大利亚沃尔特和伊丽莎·霍尔医学研究所)、Michael I. Love(美国北卡罗来纳大学教堂山分校)、Anthony T. Papenfuss(澳大利亚沃尔特和伊丽莎·霍尔医学研究所)
发表时间: 2024-06-14
发表期刊: Nature Methods
影响因子: 36.1
DOI: 10.1038/s41592-024-02299-2
摘要
组学数据的增长给数据操作、分析和整合带来了不断变化的挑战。为了应对这些挑战,Bioconductor 提供了一个广泛的社区驱动的生物数据分析平台。同时,tidy R 编程提供了革命性的数据组织和操作标准。在这里,作者展示了 tidyomics 软件生态系统,将 Bioconductor 与 tidy R 范式连接起来。该生态系统旨在简化组学分析、简化学习并鼓励跨学科合作。作者通过分析人类细胞图谱中的 750 万个外周血单核细胞,涵盖六个数据框架和十个分析工具,证明了 tidyomics 的有效性。
前言
基因组学、表观基因组学、转录组学、空间分析和多组学的高通量技术彻底改变了生物医学研究,为数据操作、探索、分析、整合和解释带来了机遇和挑战。为了应对这些挑战,科学界开发了用于数据组织和专门操作的面向对象框架。
为了应对软件领域的复杂性,Bioconductor 已成为用于组学数据分析的首要 R 软件存储库和平台。Bioconductor 为数据处理工作流程和统计分析提供国际标准化和互操作性。凭借广泛的注释资源和链接元数据的标准化数据格式,Bioconductor 促进了可重复性和社区驱动的开源开发。
最近,tidy R 范式和 tidyverse 软件生态系统通过优先考虑直观的数据表示和操作而不是复杂的数据结构和语法,改变了基于 R 的数据科学。该范式使用表格来表示数据,将变量作为列,将观察结果作为行。它通过使用标准化和自然语言词汇的管道中连接的操作来简化数据操作。tidyverse 的组件被列为最常下载的 R 包,并在全球数据科学和生物信息学项目中广泛教授。
Bioconductor 在很大程度上仍然独立于 tidyverse 生态系统。通过提供标准数据格式和分析的 tidy 接口,在这两个生态系统之间架起一座桥梁,将使研究人员能够将注意力从技术挑战转移到生物学问题。此外,利用数据科学教育标准将降低分析不同组学数据的进入门槛。
研究结果
1. tidyomics 生态系统概览
在这篇 Brief Communication 中,作者介绍了 tidyomics,这是一个可互操作的软件生态系统,它将 Bioconductor 和其他组学分析框架(例如 Seurat)与 tidyverse 连接起来。该生态系统可以通过 Bioconductor 中提供的单个 meta-package 进行安装。tidyomics 包括三个新软件包:tidySummarizedExperiment、tidySingleCellExperiment、tidySpatialExperiment,以及五个公开可用的 R 软件包:plyranges、nullranges、tidyseurat、tidybulk、tidytof。重要的是,虽然 tidyomics 以整齐的格式表示组学数据(Fig. 1a),但它保持原始数据容器和方法不变,确保与现有软件的兼容性、可维护性和长期 Bioconductor 支持。
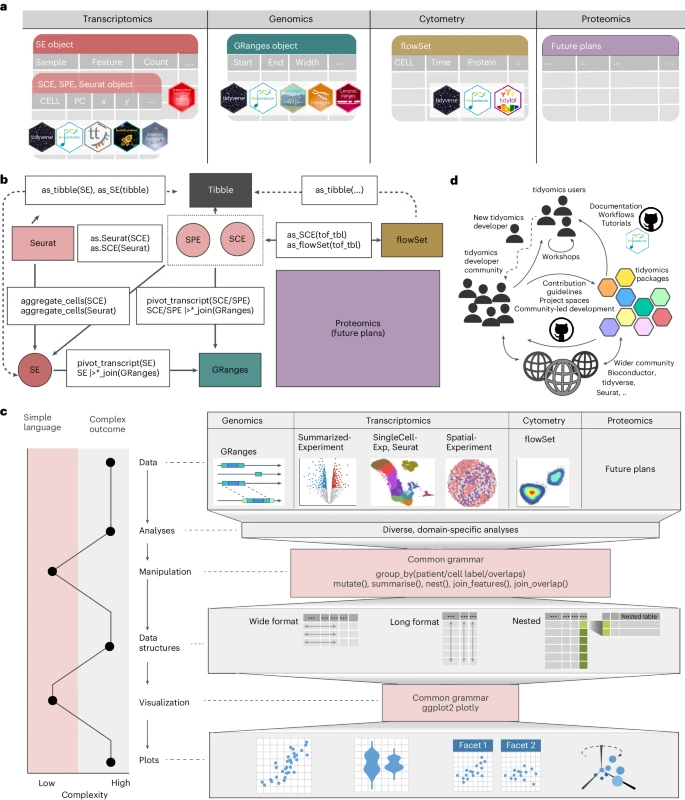
a. 数据接口图显示了不同数据容器的一致数据表示。六边形图标代表每个数据容器的兼容 R 包。
b. R/Bioconductor 中丰富数据对象的景观,以 tidyomics 动词作为连接这些对象的路径。数据容器由圆角矩形和将它们连接为白框的函数表示。SPE, SpatialExperiment; SCE, SingleCellExperiment; SE, SummarizedExperiment。
c. 左:简洁的 tidy 语法与复杂的结果和输入数据容器之间的对比。右图:示例工作流程包括数据、生物分析、数据/结果操作和总结、不同的数据结构、可视化和结果图。粉色区域包括跨组学共享语法的基础设施。
d. tidyomics 社区的参与是多方面的,以一套专为简化数据分析而定制的 R 包为中心。
在 Bioconductor 中,GenomicRanges 将基因组特征组织为行中的范围(例如,基因、外显子、单核苷酸多态性 (SNP) 和 CpGs),并与列中的变量(例如,范围宽度)链接(如 BED 格式)。plyranges 将 dplyr 动词扩展到 GenomicRanges 对象,促进范围整合、重叠分析、汇总和可视化。plyranges 与 nullranges 交互以匹配或 bootstrapping 范围以执行重叠富集分析。
在 Bioconductor 中,SummarizedExperiment 和 SingleCellExperiment 将转录丰度组织为与 metadata 链接的特征/基因样本矩阵。tidyomics 通过提供一个表格界面来概括变量的概念,其中观察值(例如,gene-sample pair)作为行,变量(例如,abundance 和 metadata)作为列。这种方法可以使用 tidyverse 进行复杂的过滤、汇总、分析和可视化。tidybulk 为 bulk 和 pseudobulk 数据提供了整洁且模块化的分析管道。
Bioconductor 的 flowCore 软件包将来自质量、流式和基于序列的细胞计数的数据组织在逐个特征矩阵中,并促进数据操作。tidytof 将 flowCore 与 tidyverse、tidySingleCellExperiment 和 tidySummarizedExperiment 连接起来。
Bioconductor 的 SpatialExperiment 组织来自基于细胞或像素的技术的数据,例如 10x Genomics Xenium、CosMX、Mibi 和 MERSCOPE。tidySpatialExperiment 为具有空间坐标的数据提供了一个 tidy 接口,并提供了专门的操作,例如基于几何和手绘形状的门控。
tidyomics 是一个统一且可互操作的组学技术软件生态系统,涵盖多个组学分析框架。通过转换和连接操作,功能网络连接所有数据容器(Fig. 1b)。这种统一的方法有助于无缝容器切换,减少对由特定领域语法创建的特定框架的依赖,并有效地增加所用工具的范围(Fig. 1c)。
2. tidyomics 生态系统的表现
为了证明 tidyomics 的实用性和可扩展性,作者测试了 750 万个血细胞的外周免疫系统的性别转录组差异。作者的生态系统无缝地连接了六个数据和分析框架(Fig. 2a),展示了一致使用 tidy R 语法而不是混合 base R、DuckDB、Seurat、SingleCellExperiment、SummarizedExperiment、DGEList、GenomicRanges 语法的好处。预处理后,作者使用多级差异表达模型测试了 26 种免疫细胞类型的 15,494 个 pseudobulk 样本(Fig. 2b)。作者发现 T CD4 naive cells、T effectors cells、B memory cells 在性别之间变化最大(Fig. 2c)。大多数与性别相关的转录变化(不包括性染色体)是细胞类型特异性的,而不是共享的(false discovery rate (FDR) <0.05)(Fig. 2d)。作者测试了对 CD4 naive cells 中的性别或其与年龄相互作用具有显着影响 (FDR<0.05) 的基因与三种免疫细胞相关和性别偏见疾病的全基因组关联研究 (GWAS) SNPs 的接近程度:硬化症、类风湿性关节炎和系统性红斑狼疮(Fig. 2e)。作者发现九个基因与这些疾病相关的 SNPs 重叠或接近(IL2RA、CD40、KCP 与两种或多种疾病相关)。很大一部分性别相关基因(41%)定义了不同的免疫衰老轨迹(Fig. 2f)。

a. tidyomics 支持大规模跨框架分析。logos 代表数据和分析框架。连接线代表管道,按对象类型着色。平行线代表并行工作流程。
b. Pseudosample UMAP,按细胞类型着色。NK, natural killer。
c. 按性别从最大到最小变化的细胞类型排名,按 b 着色。
d. 性别影响或其与年龄相互作用的前九种细胞类型之间存在大量基因重叠。
e. CD4 naive cells 中的性别相关基因与多发性硬化症、类风湿性关节炎和系统性红斑狼疮的 GWAS SNPs 的重叠。MS,多发性硬化症;RA,类风湿性关节炎;SLE,系统性红斑狼疮。
f. 与年龄无关的性别效应或性别-年龄相互作用的显着(FDR <0.05)基因的分数。箱线图中心线代表中值,上下铰链代表第一和第三四分位数。下须线从下铰链延伸至四分位距或最低值的 1.5 倍。上须线从上铰链延伸至四分位距或最高值的 1.5 倍。
g. 标准编程和 tidyverse 编程之间的代码可读性比较。展示的两项任务是可视化基因组距离直方图(左)和根据单细胞数据计算多基因特征(右)。
h. tidyomics 生态系统与标准(non-tidy)编程相比的变量、代码行和时间效率的基准测试。这些操作包括每个包的常见操作和分析 (Methods)。
此分析表明 tidyomics 允许跨不同数据源重新利用代码。例如,可以使用模块化、一致的语法组装成紧凑且清晰的管道来执行基因组和转录组数据的复杂操作和可视化(Fig. 2g)。与标准对应物相比,更少的中间变量和代码行提高了易读性和编程简单性,同时不会产生重大的执行时间开销(Fig. 2h)。Tidy R 支持函数式编程(例如,向量化而不是 for 循环),这有助于避免变量更新引起的错误,尤其是在交互式编程中。
讨论
作者专门的开发者社区采用的 Bioconductor 编程标准和贡献指南为生态系统的长期可维护性奠定了坚实的基础(Fig. 1d)。这个生态系统将会不断发展,包括具有兼容目标和来自 Bioconductor 和 CRAN 标准的 R 包。虽然 tidyomics 目前专注于简化和协调,但新颖的分析和操作工具也是其目标之一。
tidyverse 和 Bioconductor 生态系统正在改变基于 R 的数据科学和生物数据分析。tidyomics 弥合了这些生态系统之间的差距,使分析师能够在组学分析中利用 tidy 数据原则的力量。这种整合促进了跨学科合作,减少了新用户的进入障碍,并增强了代码的可读性、可重复性和透明度。适用于生物软件的 tidy 标准创建了一个可扩展的开发生态系统,独立研究人员可以在其中与新软件交互。最终,由新的、公开可用的 R 包组成的 tidyomics 生态系统有可能大大加速科学发现。
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布