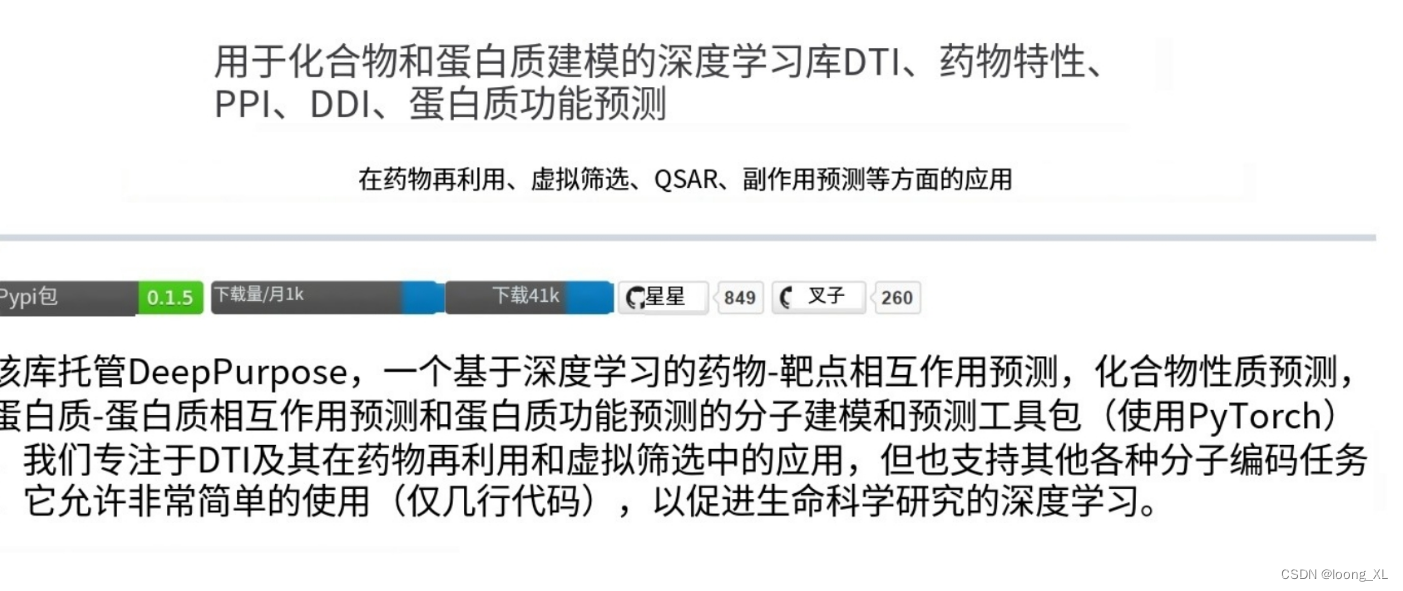
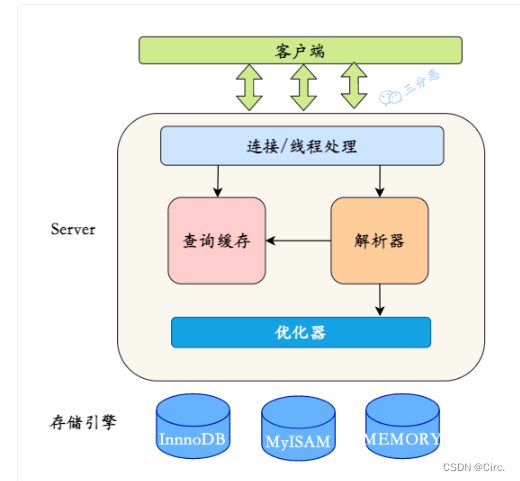
ColdDTA发表在Computers in Biology and Medicine 的一篇一区文章
突出
•
数据增强和基于注意力的特征融合用于药物靶点结合亲和力预测。
•
与其他方法相比,它在 Davis、KIBA 和 BindingDB 数据集上显示出竞争性能。
•
可视化模型权重可以获得可解释的见解。
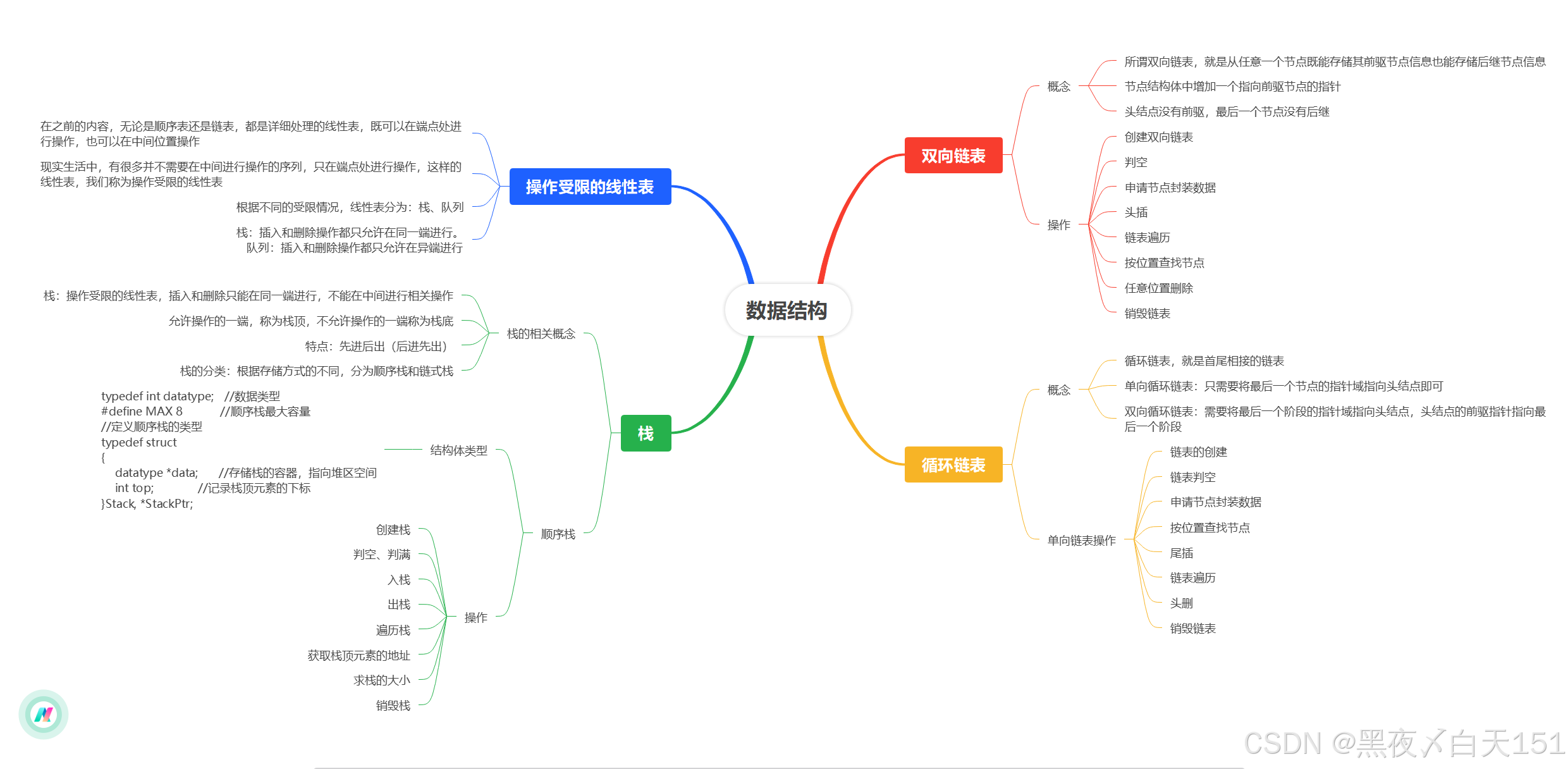
文章目录
前言
提示:这里可以添加本文要记录的大概内容:
例如:随着人工智能的不断发展,机器学习这门技术也越来越重要,很多人都开启了学习机器学习,本文就介绍了机器学习的基础内容。
提示:以下是本篇文章正文内容,下面案例可供参考
一、摘要
药物靶点亲和力(DTA)的准确预测在药物发现和开发中起着至关重要的作用。最近,深度学习方法在随机拆分的公共数据集上显示出优异的预测性能。然而,仍然需要对这种拆分方法进行验证,以反映实际应用中的实际问题。在冷启动实验设置中,测试集中的药物或蛋白质不会出现在训练集中,深度学习模型的性能通常会显着下降。这表明提高模型的泛化能力仍然是一个挑战。为此,在本研究中,我们提出了ColdDTA:利用数据增强和基于注意力的特征融合来提高预测药物-靶标结合亲和力的泛化能力。具体来说,ColdDTA通过去除药物的子图来生成新的药物-靶标对。基于注意力的特征融合模块也用于更好地捕捉药物-靶点的相互作用。我们在三个基准数据集上进行了冷启动实验,Davis 和 KIBA 数据集上的一致性指数 (CI) 和均方误差 (MSE) 结果表明,ColdDTA 优于五种最先进的基线方法。同时,BindingDB数据集上的受试者工作特征下面积(ROC-AUC)结果表明,ColdDTA在分类任务上也具有更好的性能。此外,可视化模型权重可以获得可解释的见解。总体而言,ColdDTA可以较好地解决现实的DTA预测问题。该代码已向公众开放。
二、引言
药物开发是一个漫长而昂贵的过程。根据调查,开发一种新药大约需要13年时间,成本为0.6-26亿美元,成功率不到10%[1]。这促使人们使用计算方法来降低成本和提高效率。药物分子与具有适当化学特性和亲和力的靶标结合的过程是治愈疾病的关键标准之一。因此,准确预测药物-靶标结合亲和力对于药物发现至关重要[2]。
使用计算机进行药物靶标亲和力(DTA)预测的方法可分为基于结构的方法、基于配体的方法和混合方法[3]。分子对接是最早和使用最广泛的基于结构的方法之一。然而,这种方法需要蛋白质的3D结构和较长的计算时间,即使是现在,仍然有大量未知的蛋白质结构。尽管AlphaFold2能够预测大多数蛋白质[4],但并非所有结果都非常准确[5]。基于配体的方法假设具有相似化学性质的药物分子具有相同的化学活性,因此可以与相似的靶标相互作用。然而,当已知配体数量不足时,基于配体的方法的预测结果往往变得不可靠。混合方法有望克服上述局限性。其中之一,传统的机器学习方法,如支持向量机(SVM)和随机森林(RF),需要特征工程来提取药物和蛋白质特征[6],特征工程非常耗时,并且会因信息丢失而影响预测性能。近年来,已经开发了几种深度学习方法,如DeepDTA [7]和GraphDTA [8],用于药物-靶标相互作用预测。
一些深度学习模型使用基于序列的输入数据,如WideDTA [9],用简化分子输入线输入系统(SMILES)表示药物,用氨基酸序列表示蛋白质,然后通过卷积神经网络(CNN)或递归神经网络提取特征(RNN)。
但这种方法缺乏分子拓扑结构。
为了弥补这一不足,许多方法将药物视为图结构,化合物的原子和化学键对应于图的顶点和边缘,然后通过图神经网络(GNN)进行特征提取。DeepGS [10]、MolTrans [11]和MgraphDTA [12]都以图表示药物,并在一些公共数据集上表现出优异的预测性能。同样,一些研究也关注蛋白质在药物靶标结合过程中的作用,其中 DrugVQA [13]、AttentionSiteDTI [14] 和 GSAML-DTA [15] 使用接触图来表示蛋白质,实验结果表明,使用蛋白质的 3D 结构可以做出更好的预测。Transformer [16] 也被用于预测药物-靶标相互作用。Chen等人提出的TransformerCPI模型将药物-靶标相互作用视为二元分类问题,并使用编码器-解码器架构进行模型训练,最终预测两者是否可以相互作用[17]。
一些深度学习模型使用基于序列的输入数据,如WideDTA [9],用简化分子输入线输入系统(SMILES)表示药物,用氨基酸序列表示蛋白质,然后通过卷积神经网络(CNN)或递归神经网络提取特征(RNN)。但这种方法缺乏分子拓扑结构。为了弥补这一不足,许多方法将药物视为图结构,化合物的原子和化学键对应于图的顶点和边缘,然后通过图神经网络(GNN)进行特征提取。DeepGS [10]、MolTrans [11]和MgraphDTA [12]都以图表示药物,并在一些公共数据集上表现出优异的预测性能。同样,一些研究也关注蛋白质在药物靶标结合过程中的作用,其中 DrugVQA [13]、AttentionSiteDTI [14] 和 GSAML-DTA [15] 使用接触图来表示蛋白质,实验结果表明,使用蛋白质的 3D 结构可以做出更好的预测。Transformer [16] 也被用于预测药物-靶标相互作用。Chen等人提出的TransformerCPI模型将药物-靶标相互作用视为二元分类问题,并使用编码器-解码器架构进行模型训练,最终预测两者是否可以相互作用[17]。
尽管一些深度学习方法在DTA问题上表现出出色的预测性能,但这些方法大多是在随机拆分的数据集上进行评估的(测试集中的药物和目标已经出现在训练集中),这可能导致信息泄露,使结果过于乐观[18]。从应用的角度来看,大多数蛋白质或药物不会出现在训练集中[19]。当数据集根据更现实的场景进行拆分时,许多模型的预测性能急剧下降[20],有时预测性能比传统机器学习方法更差[21]。这表明当前DTA模型的泛化能力仍有待提高。
为了提高模型的泛化能力,使其在真实场景中发挥更大的作用,我们首先考虑了数据增强方法的运用。数据增强方法已广泛应用于计算机视觉[[22],[23],[24]]和自然语言处理[[25],[26],[27]],并已被证明可以有效提高模型泛化。还提出了多种有效的数据增强方法来预测分子特性[28,29]。然而,据我们所知,数据增强尚未用于提高使用分子图表示药物的 DTA 预测方法的泛化性能。这可能是由于生物化学告诉我们,化合物中原子的微小变化也可能导致分子物理和化学性质的显着变化,以及小药物分子与靶标之间结合亲和力的变化。如图1所示,对于邻苯二酚分子,去除一些原子为苯酚会将其水溶性从溶于水变为微溶于水。另一方面,特征融合是指来自不同层或分支的特征的组合,在现代网络架构中无处不在,使用合理的特征融合方法可以有效提高模型性能。但现有的大多数DTA预测方法只是简单地将药物和蛋白质的特征串联起来,进行最终结果预测,这限制了药物与靶点之间的相互作用,限制了模型的预测和泛化性能。
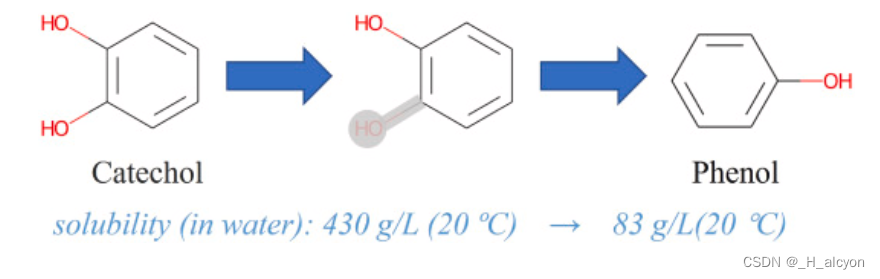
图 1.影响其性质的原子变化的图示。
在这项工作中,我们提出了一种称为ColdDTA的药物靶标亲和力训练方法,该方法使用数据增强和基于注意力的特征融合来预测药物和蛋白质之间的结合亲和力。具体来说,在数据处理阶段,将一定比例的药物子图移除,并与原始目标构成新的数据对,然后在训练阶段使用。此外,ColdDTA使用药物-蛋白质特征融合模块来取代简单的特征串联。通过对两个基准数据集进行实验,我们发现与其他方法相比,ColdDTA的整体性能最佳。我们的消融实验结果也证明了数据增强方法和药物-靶点融合模块的有效性。本研究的主要贡献总结如下:
1.
采用一种新的数据增强方法,通过去除药物的子图,与原目标形成新的数据对,可以有效提高模型的泛化能力。
该文提出一种基于注意力的药物与靶点数据融合方法,该方法能够更好地将药物特征与蛋白质特征融合,有利于模型进行亲和值预测。
在两个公开数据集上进行了大量实验,验证了在接近真实实验环境的数据集设置下,ColdDTA的预测能力与基线模型相比有显著提高。此外,实验还证明了数据增强和药物靶点融合的有效性。
三、方法
1.概述
我们提出了一种称为ColdDTA的端到端训练方法来提高DTA预测性能。我们对 DTA 问题的定义如下:让
是一批标记的数据,其中
是小分子药物的SMILES表示,
是蛋白质,并且
是通过实验获得的结合亲和力值。模型的整体训练过程如图 2 所示。它由数据增强模块、药物特征提取模块、蛋白质特征提取模块、药物-靶点融合模块和预测模块五部分组成。
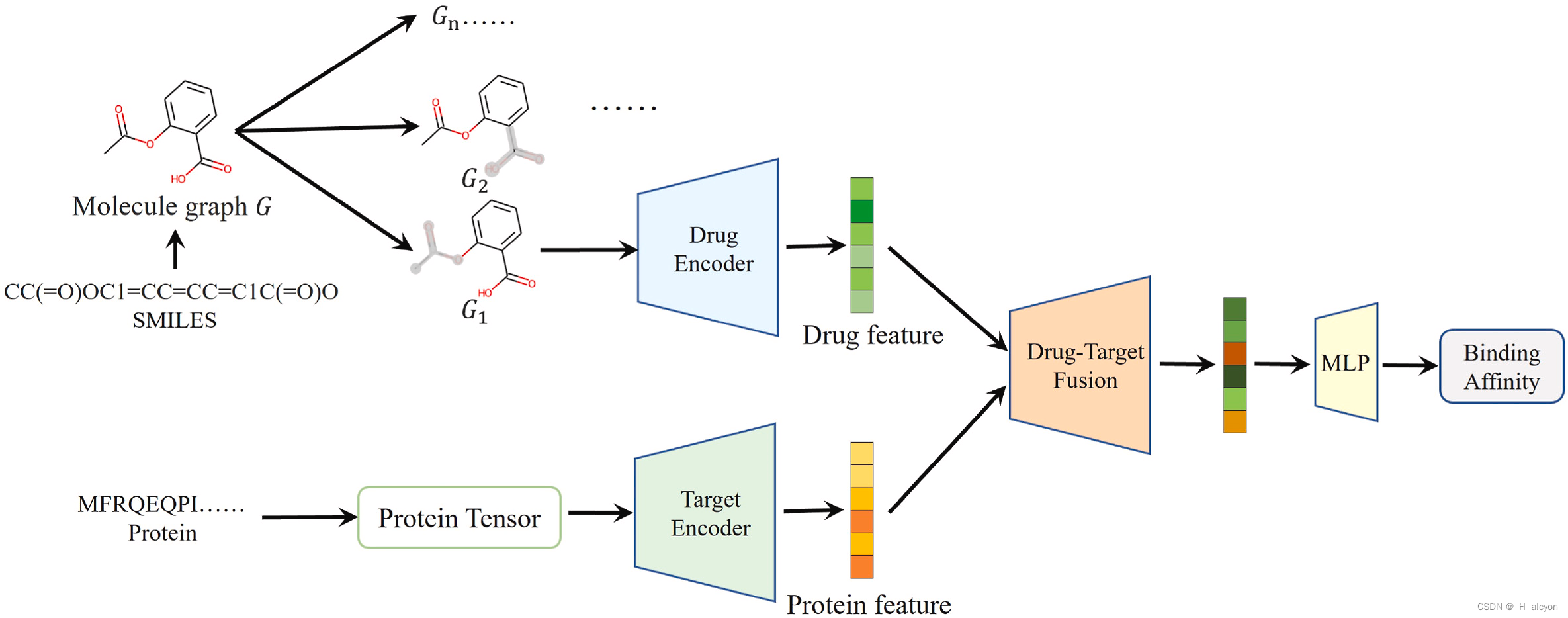
图 2.拟议的 ColdDTA 的框架。首先,去除药物的子图,并与原始蛋白质形成新的对。其次,利用GNN和CNN分别提取分子图和蛋白质的特征;第三,将上一步的特征输入到药物-靶点融合模块进行交互,以获得更有效的特征表示。最后,将融合特征输入MLP以预测结合亲和力。
2.数据增强策略
数据增强是提高神经网络数据效率、增强泛化性能的必要条件[30]。使用数据增强方法可以提高模型的预测性能似乎很明显,但很少有研究使用这种策略来预测药物-靶点相互作用预测问题,因此我们在DTA预测中引入了一种新的数据增强策略。具体来说,在药物-靶点配对中,我们首先随机选择分子图中的一个原子作为起始节点,然后去除起始节点并递归地去除其相邻节点,直到去除一定比例的子图,而相应的靶点和结合亲和力保持不变。在这个过程中,原子基团的去除是完全随机的,这类似于分子性质预测的对比学习[31]。图 3 显示了数据增强过程的输出。在训练阶段,一对药物化合物和靶蛋白通过去除子图产生多个新对。需要注意的是,在测试阶段不会删除任何子图。
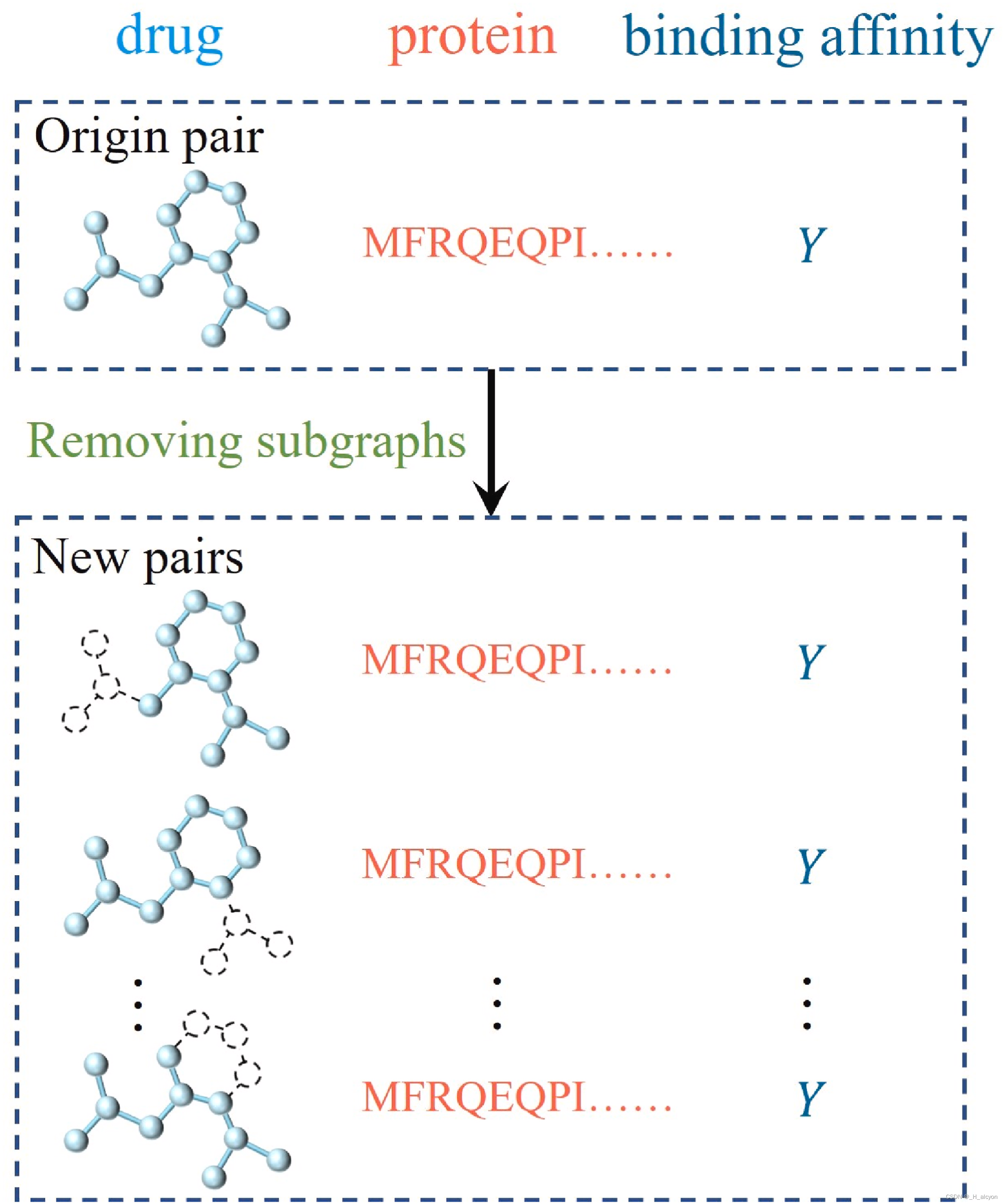
3.药物特征提取模块

4.蛋白质特征提取模块

5.药物-靶点融合模块

代码如下(示例):
data = pd.read_csv(
'https://labfile.oss.aliyuncs.com/courses/1283/adult.data.csv')
print(data.head())
该处使用的url网络请求的数据。
总结
提示:这里对文章进行总结:
例如:以上就是今天要讲的内容,本文仅仅简单介绍了pandas的使用,而pandas提供了大量能使我们快速便捷地处理数据的函数和方法。