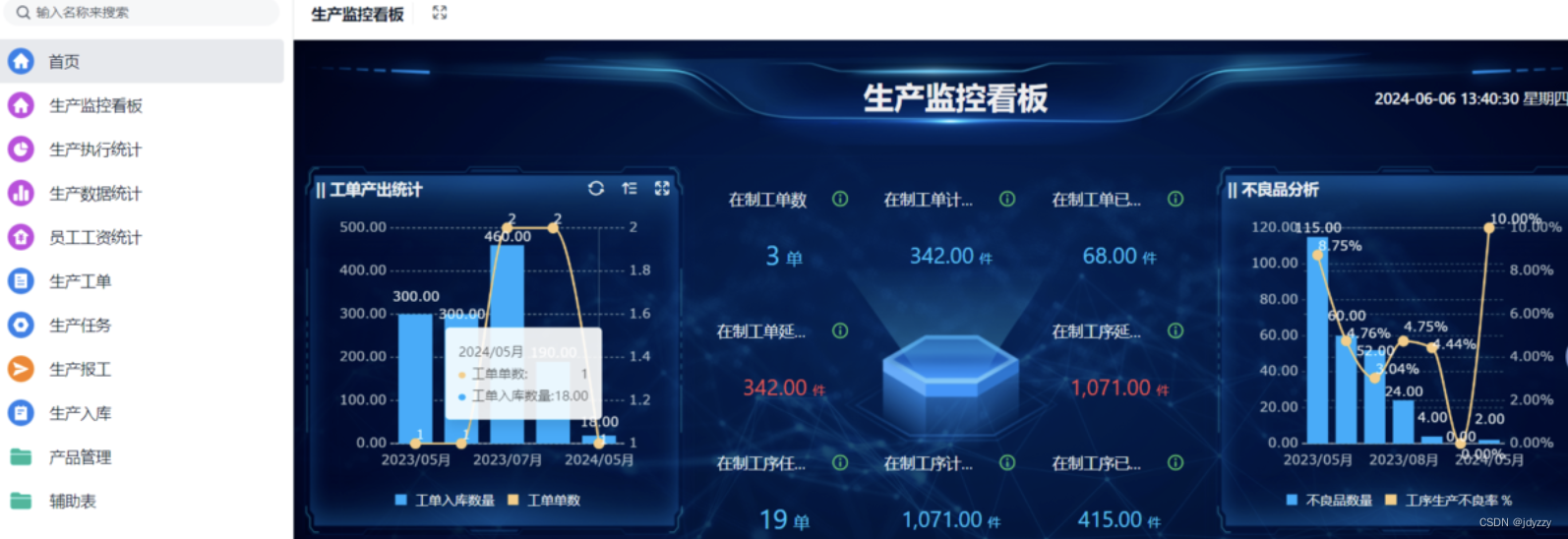
在一个完整的分子动力学模拟中,一般包括以下几个步骤:
1.选择将要使用的力场,并根据模拟体系确定力场参数,构建力场文件;
2.产生初始构型,搭建模拟体系;
3.模拟退火(simulated annealing);
4.平衡体系;
5.模拟数据采样;
6.数据分析、处理。
产生初始构型经验总结
1.产生初试构型的一般步骤是:
1) 给出分子内原子坐标;
2) 按照某种分布将分子安放到空间不同位置(可以按照某种晶格排列,对于液体、气体,亦可随排列);
2.分子内原子位置排布不能偏离平衡位置太远,否则,可能会因为能量或力发散,导致模拟出错;
3.如果分子比较复杂,可以先跑一个几皮秒的单分子模拟,再取其构型作为平衡的分子构型;
4.分子间位置可以偏离平衡位置,但应注意避免原子位置重叠的情况,这样同样会导致能量发散的问题,所以,初试构型亦可让分子距离远一点;
5.不要让原子超出你给定的盒子大小;
模拟退火经验总结
1.模拟退火过程需要根据你的要求和体系的性质来设计,目的是加速体系达到平衡;
2.如果需要达到较好的平衡效果,则可以选取更为细致的温度梯度,使体系温度缓慢降低;另外,为了提高效率,高温的模拟时间可以较短,温度越低,所需的平衡时间越长;
3.如果进行高温退火(沸点以上),则需要进行NVT模拟,这时第一步是确定,体系的盒子大小;
4.盒子大小可以从实验中得到;如果缺少实验数据,则需要通过NPT模拟来计算,虽然体积的平衡较快,但由于初始构型偏离平衡态较远,这一步定出的体积可能不准;可以待模拟退火结束后,在进行NVT模拟(数据采样)前,再通过NPT模拟准确的定出平衡态盒子大小;
5.初始构型给出的体系大小往往大于平衡态的体系大小,这时可以跑一个高压(eg.10000 atm)的NPT模拟,让体系快速压缩;
6.在平衡过程(退火)中,建议选取berendsen热耦,不建议选择Nose-Hoover热耦,后则较为细致,在远离平衡时,可能会出现较大的温度涨落;
一、GROMACS分子动力学蛋白模拟、药物开发溶剂筛选
1 分子模拟基础理论
1.1 统计力学理论概述
1.2 主要算法介绍:最速下降法、共轭梯度法、有限差分法
1.3 力场、力场类型、参数和分类:AMBER、CHARMM、MMX、CVFF、OPLS
1.4 基础知识:积分迭代器、积分步长选取、温度控制、压力控制、周期性边界条件
1.5 模拟基本流程:能量最小化、NVE弛豫、NVT控温、NPT控压、MD平衡模拟
1.6 计算化学基本概念:范德华表面、分子表面、接触表面、溶剂可及表面、势能面
2 GROMACS程序入门——学会编译方法,安装自己的GROMACS可执行程序,并运行一个例子。
2.1 版本/安装/运行
•Linux入门操作及方法 •并行介绍和环境搭建 •win版、linux版编译安装及运行 •win版下使用linux系统编译安装及运行GROMACS程序
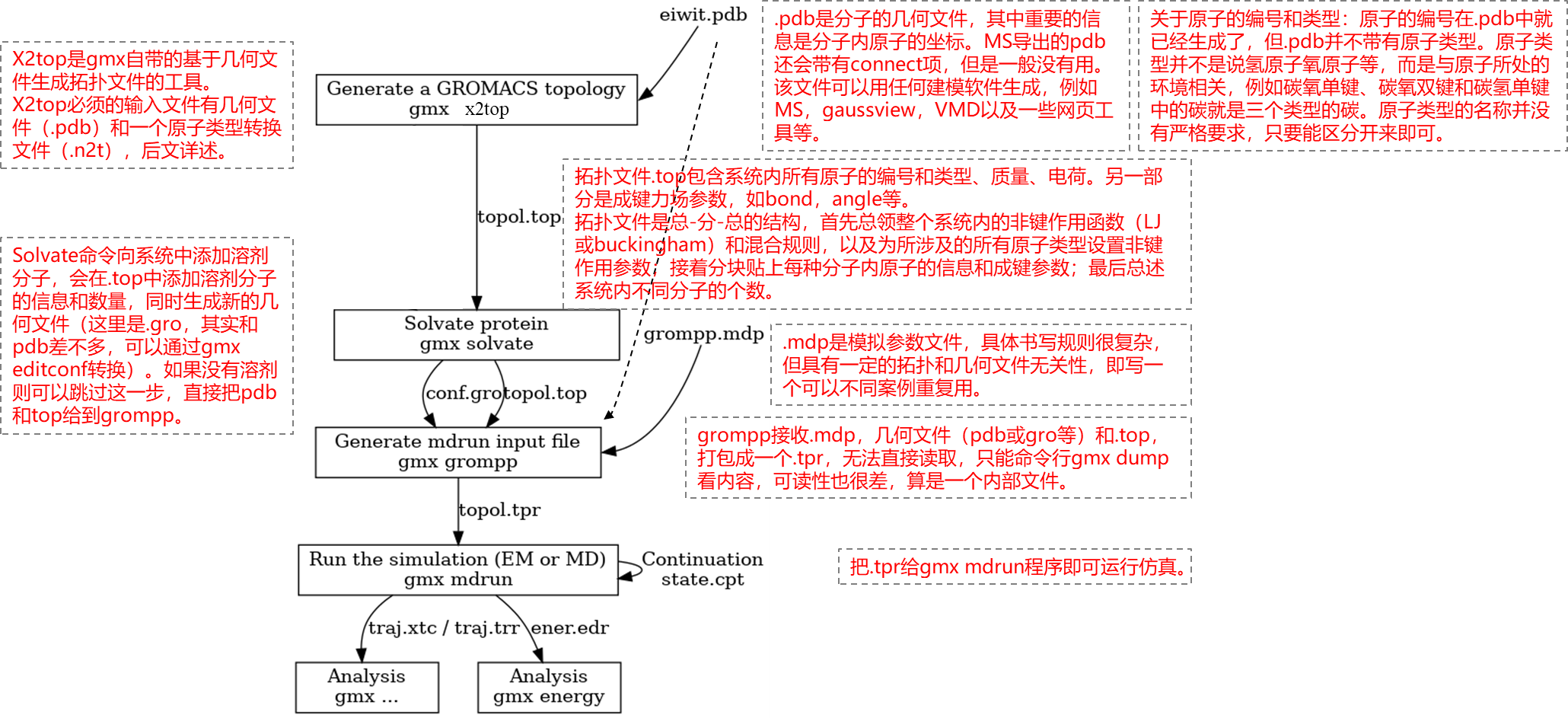
2.2 各种文件介绍:PDB、GRO、TOP/ITP、XVG、MDP
2.3力场概念、分类及力场参数修改:——探究力场具体形式,为以后创建自己体系做准备,以OPLS为例,力场的各种参数说明及修改
3 生物体系建模——掌握不同体系快速搭建方法,使学员具有扎实的建模基础
3.1 辅助工具软件:Packmol、GaussView、vmd、Grace等
3.2 模型建模/TOP文件的生成
生物小分子PDB构建、生物小分子模型及原子类型定义、结构调整(键长、键角、二面角)、生物小分子top结构构建、itp文件建立、拓扑文件生成工具
3.3 不同简单生物体系的建模
3.4 蛋白质、核酸、多肽、溶剂等复杂体系构建
3.5 构建一个简单的生物分子体系模型并运行
4 模拟结果分析——掌握不同生物体系所需分析方法,生成拓扑结构和坐标文件
4.1 模拟轨迹分析:trajectory,sasa,rdf,freevolume等
4.2 生成拓扑结构和坐标文件:editconf,genconf,pdb2gmx等
4.3 模拟能量分析:energy,enemat等
4.4 系统动态结构分析:cluster,confrms,midist等
4.5 空间分布性质:gyrate,msd,rdf,traj等
4.6 分子结构分析:hbond,order,principal,spol等
4.7 静电作用分析:dielectric,dipoles,potential等
5 水溶性蛋白质和配体作用分子动力学模拟
5.1 配体分子的处理
5.2 蛋白结构的处理
5.3 修改蛋白坐标文件
5.4 修改拓扑文件
5.5 构建盒子并放入溶剂
5.6 平衡系统电荷
5.7 能量最小化
5.8 NVT平衡
5.9 NPT平衡
5.10 模拟结果取样
6 分子动力学结果分析
6.1 轨迹文件观察
6.2 能量数据作图
6.3 计算斜方差
6.4 测量回旋半径
6.5 计算结构的RMSD值
6.6 计算原子位置的根均方波动
6.7 计算模拟过程中分子间的氢键的数目、距离或角度
7 生物膜磷脂双分子层生物膜、膜蛋白等建模
7.1 磷脂分子结构、双分子层建模
7.2 模拟水通道(蛋白、多肽等)
7.3 插入溶剂分子
7.4 模拟系统达平衡
7.5 分析溶剂分子扩散速率
7.6 分子间的氢键的数目、距离或角度
8 蛋白质结合自由能计算(伞形采样法为例)
8.1 创建一系列反应路径分子构型
8.2 提取模拟间隔质心轨迹
8.3 模拟每个构型的伞形采样
8.4 柱状图分析计算结合自由能
8.5 模拟结果讨论
9 药物分子开发溶剂筛选
9.1 介绍热力学积分方法
9.2 构建药物分子模型
9.3 建模药物分子溶解在水相,油相和醇相
9.4 计算不同相态下的分配系数
9.5 快速预测药物分配系数方法