引言
在本教程中,我们将探讨由10x Genomics公司提供的成年小鼠大脑细胞的单细胞ATAC-seq数据集。本教程中使用的所有相关文件均可在10x Genomics官方网站上获取。
本教程复现了之前在人类外周血单核细胞(PBMC)的Signac入门教程中执行的命令。我们通过在不同的系统上进行相同的分析,来展示其性能以及对不同组织类型的适用性,并提供了一个来自不同物种的示例。
创建基因活动矩阵
# compute gene activities
gene.activities <- GeneActivity(brain)
# add the gene activity matrix to the Seurat object as a new assay
brain[['RNA']] <- CreateAssayObject(counts = gene.activities)
brain <- NormalizeData(
object = brain,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(brain$nCount_RNA)
)
DefaultAssay(brain) <- 'RNA'
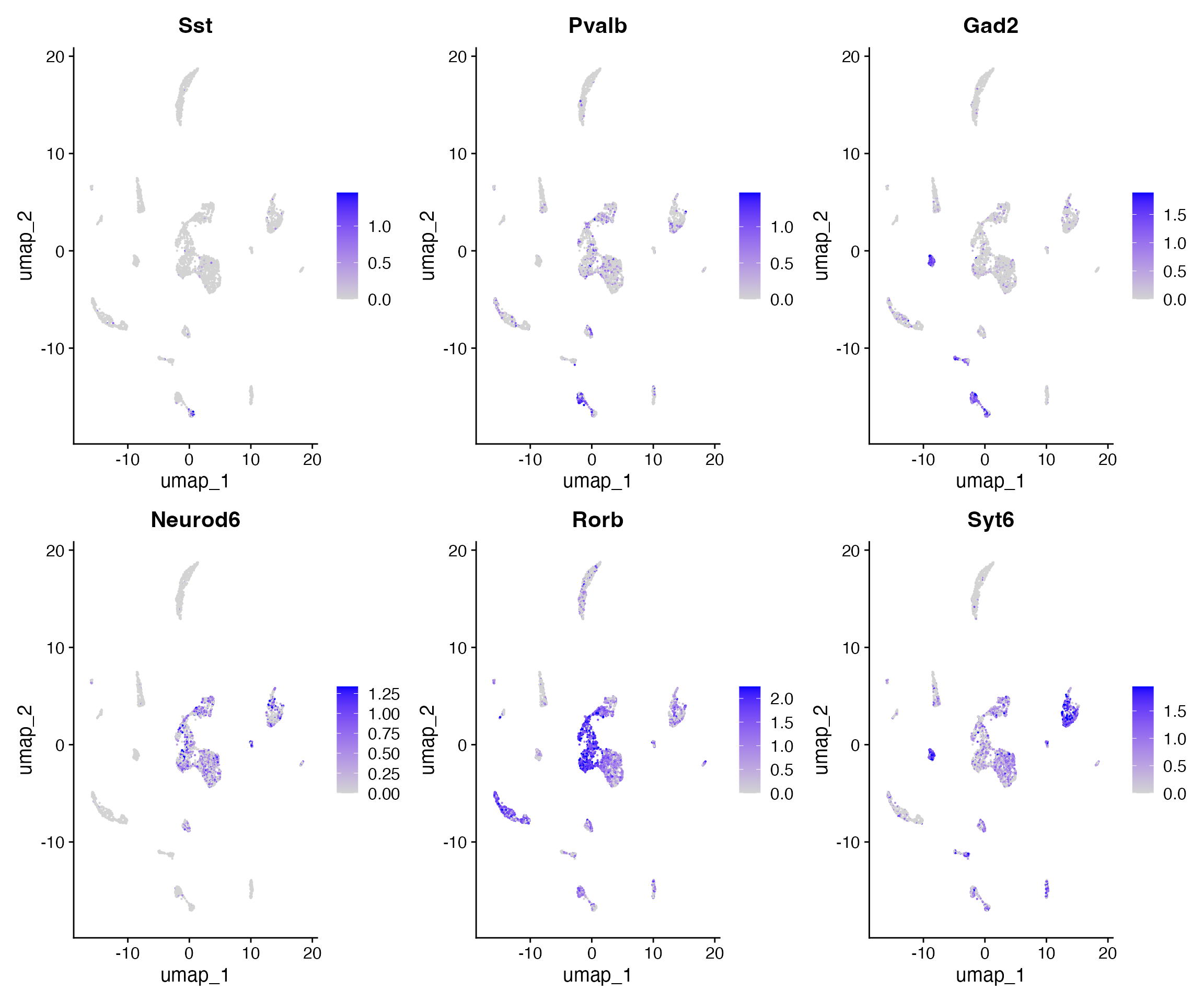
FeaturePlot(
object = brain,
features = c('Sst','Pvalb',"Gad2","Neurod6","Rorb","Syt6"),
pt.size = 0.1,
max.cutoff = 'q95',
ncol = 3
)
与 scRNA-seq 数据整合
为了更好地解读单细胞ATAC-seq数据,我们可以根据来自相同生物体系(即成年小鼠大脑)的单细胞RNA测序(scRNA-seq)实验结果,
# Load the pre-processed scRNA-seq data
allen_rna <- readRDS("../vignette_data/allen_brain.rds")
allen_rna <- UpdateSeuratObject(allen_rna)
allen_rna <- FindVariableFeatures(
object = allen_rna,
nfeatures = 5000
)
transfer.anchors <- FindTransferAnchors(
reference = allen_rna,
query = brain,
reduction = 'cca',
dims = 1:30
)
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = allen_rna$subclass,
weight.reduction = brain[['lsi']],
dims = 2:30
)
brain <- AddMetaData(object = brain, metadata = predicted.labels)
plot1 <- DimPlot(allen_rna, group.by = 'subclass', label = TRUE, repel = TRUE) + NoLegend() + ggtitle('scRNA-seq')
plot2 <- DimPlot(brain, group.by = 'predicted.id', label = TRUE, repel = TRUE) + NoLegend() + ggtitle('scATAC-seq')
plot1 + plot2
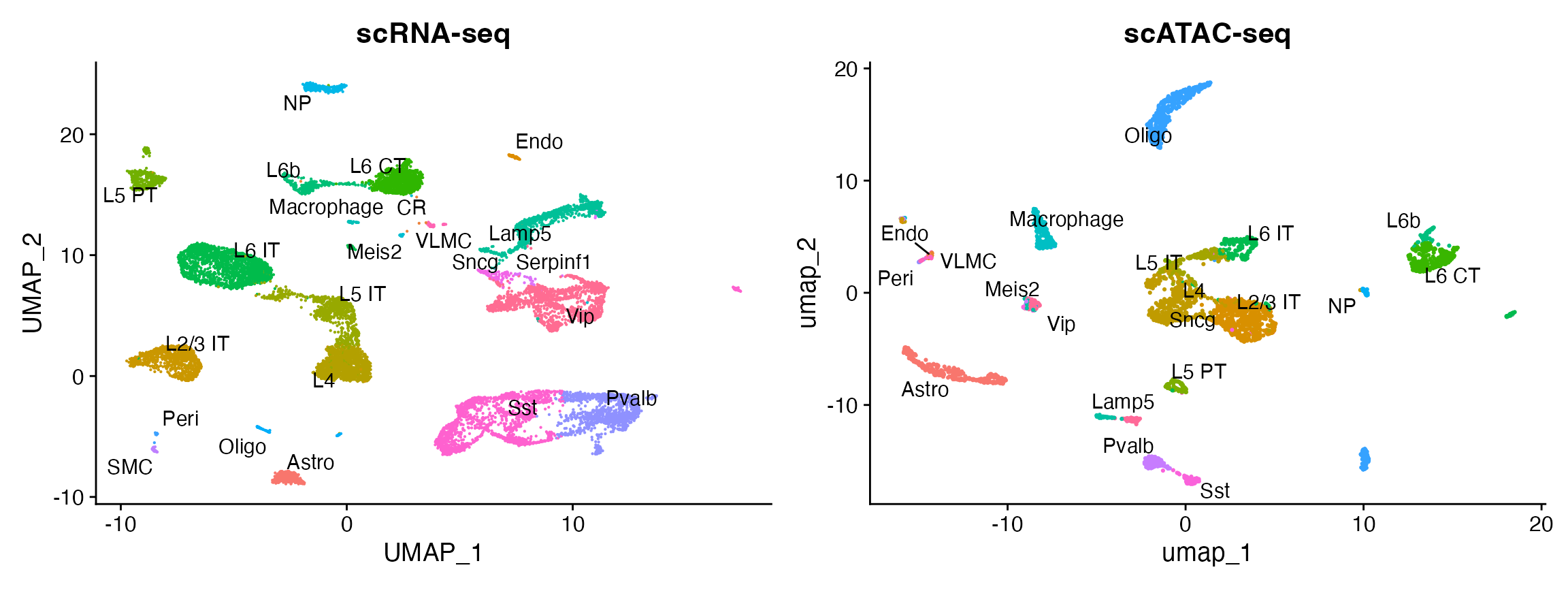
您可以看到基于 RNA 的分类与 UMAP 可视化一致,仅根据 ATAC-seq 数据计算。
查找簇之间可差异访问的峰值
在这里,我们发现皮层不同层的兴奋性神经元之间的可访问区域存在差异。
#switch back to working with peaks instead of gene activities
DefaultAssay(brain) <- 'peaks'
Idents(brain) <- "predicted.id"
da_peaks <- FindMarkers(
object = brain,
ident.1 = c("L2/3 IT"),
ident.2 = c("L4", "L5 IT", "L6 IT"),
test.use = 'LR',
latent.vars = 'nCount_peaks'
)
head(da_peaks)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
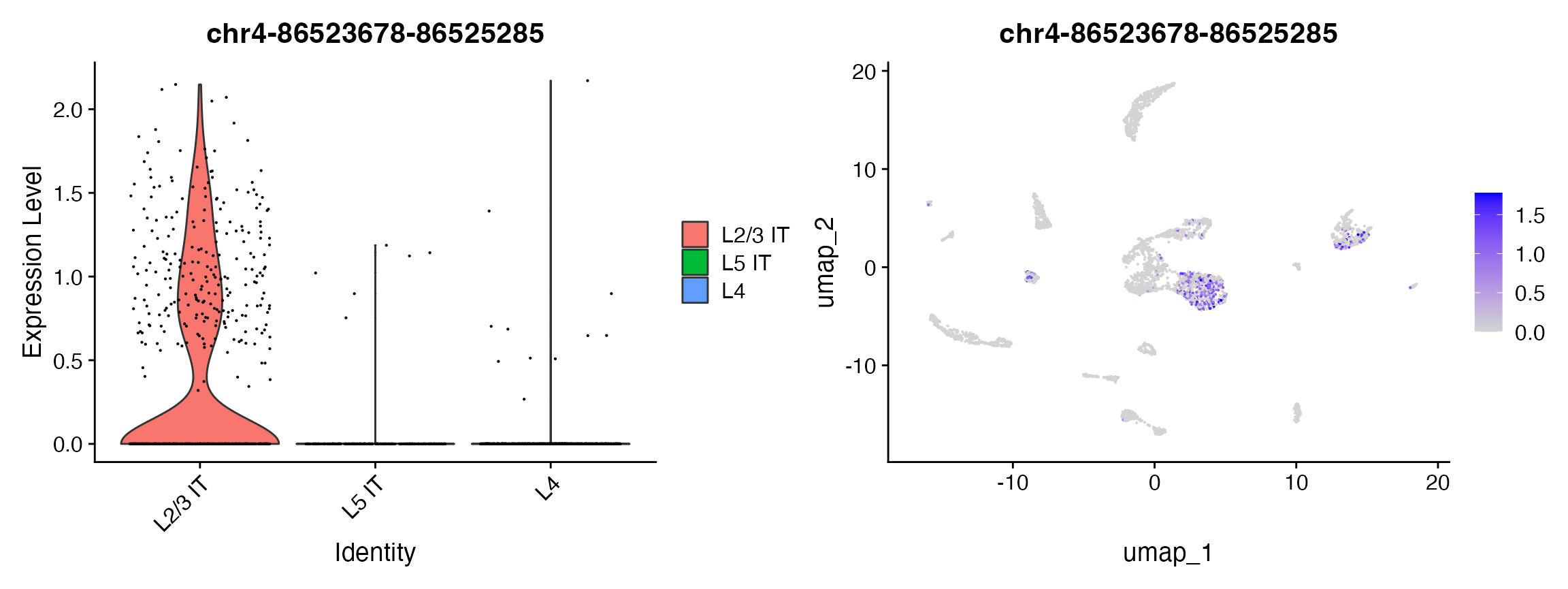
## chr4-86523678-86525285 3.266647e-69 3.691294 0.426 0.037 5.135267e-64
## chr2-118700082-118704897 8.553383e-61 2.092487 0.648 0.182 1.344617e-55
## chr15-87605281-87607659 3.864918e-55 2.450827 0.499 0.097 6.075767e-50
## chr10-107751762-107753240 1.534485e-52 1.801355 0.632 0.192 2.412257e-47
## chr4-101303935-101305131 5.949521e-51 3.427059 0.356 0.031 9.352825e-46
## chr13-69329933-69331707 1.604991e-49 -2.254722 0.140 0.435 2.523094e-44
plot1 <- VlnPlot(
object = brain,
features = rownames(da_peaks)[1],
pt.size = 0.1,
idents = c("L4","L5 IT","L2/3 IT")
)
plot2 <- FeaturePlot(
object = brain,
features = rownames(da_peaks)[1],
pt.size = 0.1,
max.cutoff = 'q95'
)
plot1 | plot2
open_l23 <- rownames(da_peaks[da_peaks$avg_log2FC > 3, ])
open_l456 <- rownames(da_peaks[da_peaks$avg_log2FC < 3, ])
closest_l23 <- ClosestFeature(brain, open_l23)
closest_l456 <- ClosestFeature(brain, open_l456)
head(closest_l23)
## tx_id gene_name gene_id
## ENSMUST00000151481 ENSMUST00000151481 Fam154a ENSMUSG00000028492
## ENSMUST00000131864 ENSMUST00000131864 Gm12796 ENSMUSG00000085721
## ENSMUST00000139527 ENSMUST00000139527 Yipf1 ENSMUSG00000057375
## ENSMUSE00001329193 ENSMUST00000185379 Gm29414 ENSMUSG00000099392
## ENSMUSE00000514286 ENSMUST00000077353 Hmbs ENSMUSG00000032126
## ENSMUST00000161356 ENSMUST00000161356 Reln ENSMUSG00000042453
## gene_biotype type closest_region
## ENSMUST00000151481 protein_coding gap chr4-86487920-86538964
## ENSMUST00000131864 lincRNA gap chr4-101292521-101318425
## ENSMUST00000139527 protein_coding cds chr4-107345009-107345191
## ENSMUSE00001329193 lincRNA exon chr1-25026581-25026779
## ENSMUSE00000514286 protein_coding exon chr9-44344010-44344228
## ENSMUST00000161356 protein_coding gap chr5-21891568-21895988
## query_region distance
## ENSMUST00000151481 chr4-86523678-86525285 0
## ENSMUST00000131864 chr4-101303935-101305131 0
## ENSMUST00000139527 chr4-107344435-107345145 0
## ENSMUSE00001329193 chr1-25008426-25009334 17246
## ENSMUSE00000514286 chr9-44345250-44346015 1021
## ENSMUST00000161356 chr5-21894051-21894682 0
head(closest_l456)
## tx_id gene_name gene_id
## ENSMUST00000104937 ENSMUST00000104937 Ankrd63 ENSMUSG00000078137
## ENSMUSE00000647021 ENSMUST00000068088 Fam19a5 ENSMUSG00000054863
## ENSMUST00000165341 ENSMUST00000165341 Otogl ENSMUSG00000091455
## ENSMUST00000044081 ENSMUST00000044081 Papd7 ENSMUSG00000034575
## ENSMUST00000070198 ENSMUST00000070198 Ppp3ca ENSMUSG00000028161
## ENSMUST00000084628 ENSMUST00000084628 Hs3st2 ENSMUSG00000046321
## gene_biotype type closest_region
## ENSMUST00000104937 protein_coding cds chr2-118702266-118703438
## ENSMUSE00000647021 protein_coding exon chr15-87625230-87625486
## ENSMUST00000165341 protein_coding utr chr10-107762223-107762309
## ENSMUST00000044081 protein_coding utr chr13-69497959-69499915
## ENSMUST00000070198 protein_coding utr chr3-136935226-136937727
## ENSMUST00000084628 protein_coding cds chr7-121392730-121393214
## query_region distance
## ENSMUST00000104937 chr2-118700082-118704897 0
## ENSMUSE00000647021 chr15-87605281-87607659 17570
## ENSMUST00000165341 chr10-107751762-107753240 8982
## ENSMUST00000044081 chr13-69329933-69331707 166251
## ENSMUST00000070198 chr3-137056475-137058371 118747
## ENSMUST00000084628 chr7-121391215-121395519 0
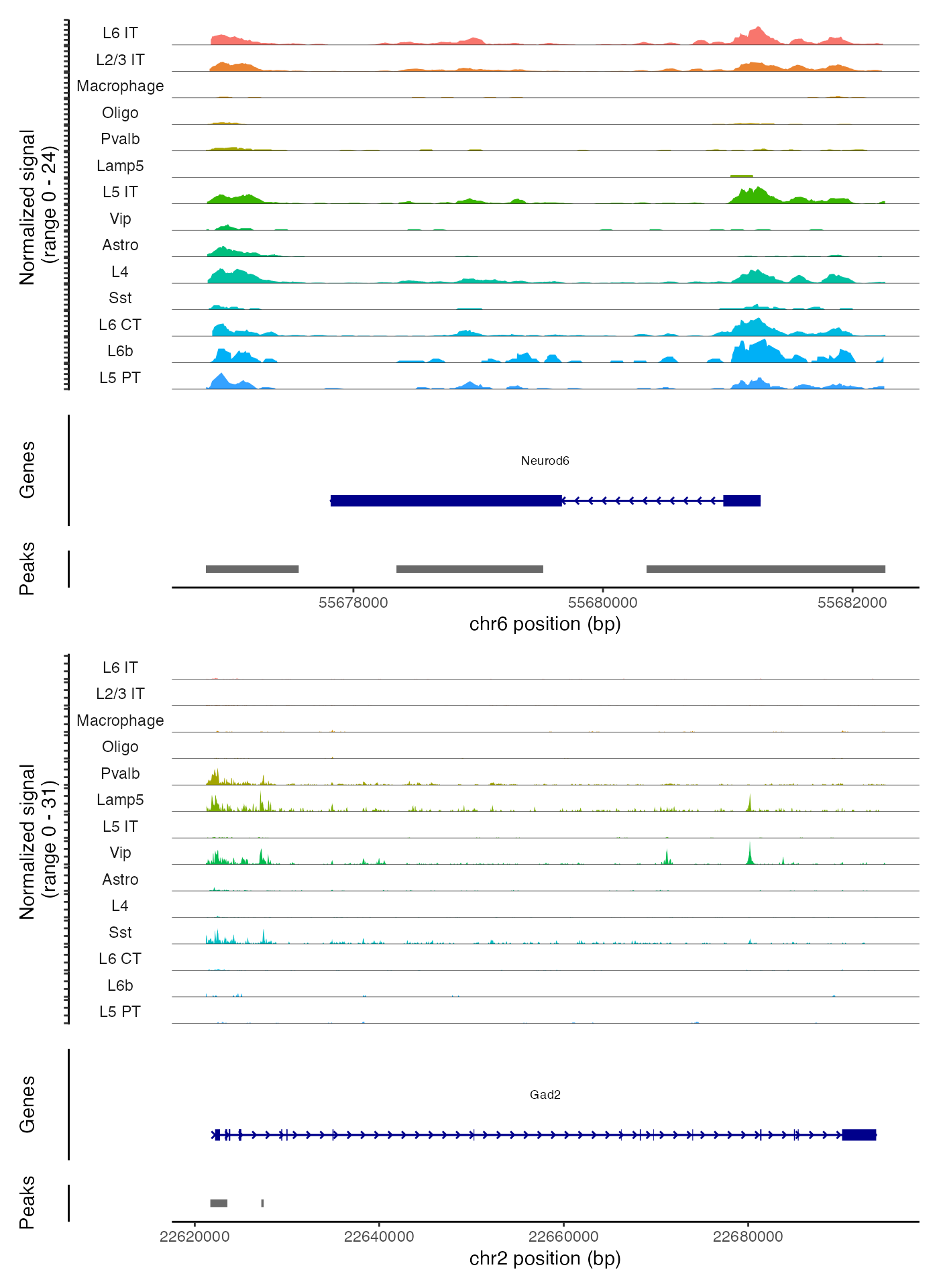
绘制基因组区域
我们同样可以利用CoveragePlot()函数,根据不同的细胞聚类、细胞类型或对象中存储的其他任何元数据信息,为特定的基因组区域绘制出分组的覆盖度图。这些覆盖度图实际上是伪批量的可访问性轨迹图,通过将同一组内所有细胞的信号进行平均,从而在视觉上展示出特定区域内DNA的可访问性情况。
# show cell types with at least 50 cells
idents.plot <- names(which(table(Idents(brain)) > 50))
CoveragePlot(
object = brain,
region = c("Neurod6", "Gad2"),
idents = idents.plot,
extend.upstream = 1000,
extend.downstream = 1000,
ncol = 1
)
本文由 mdnice 多平台发布



















![[C++初阶]string类的详解](https://img-blog.csdnimg.cn/direct/636ca1956ca746119bbb76faa50b954d.png)





![[洛谷] 刷题栈 & 队列](https://img-blog.csdnimg.cn/direct/c1798af895044f358643ffdd644b3221.png)